Summary
AlphaFold as a protein structure prediction tool developed and released by DeepMind has many possible applications in the experimental structure solution pipeline. Models from the proteome of many organisms are available for download from the AlphaFold Protein Structure Database, Developed by DeepMind and EMBL-EBI.
At Diamond we have embedded AlphaFold into our pipelines for academic users to create models specifically based on your target protein sequence. The resulting models are then used as part of the downstream processing pipelines run after data collection at Diamond.
First ensure your protein sequence (single chain only) is uploaded to ISPyB. See registering a protein for details. This should be done as early as possible, ideally prior to shipment.
AlphaFold will be run on that sequence, and if successful the results will be seen as PDB files associated to the protein:
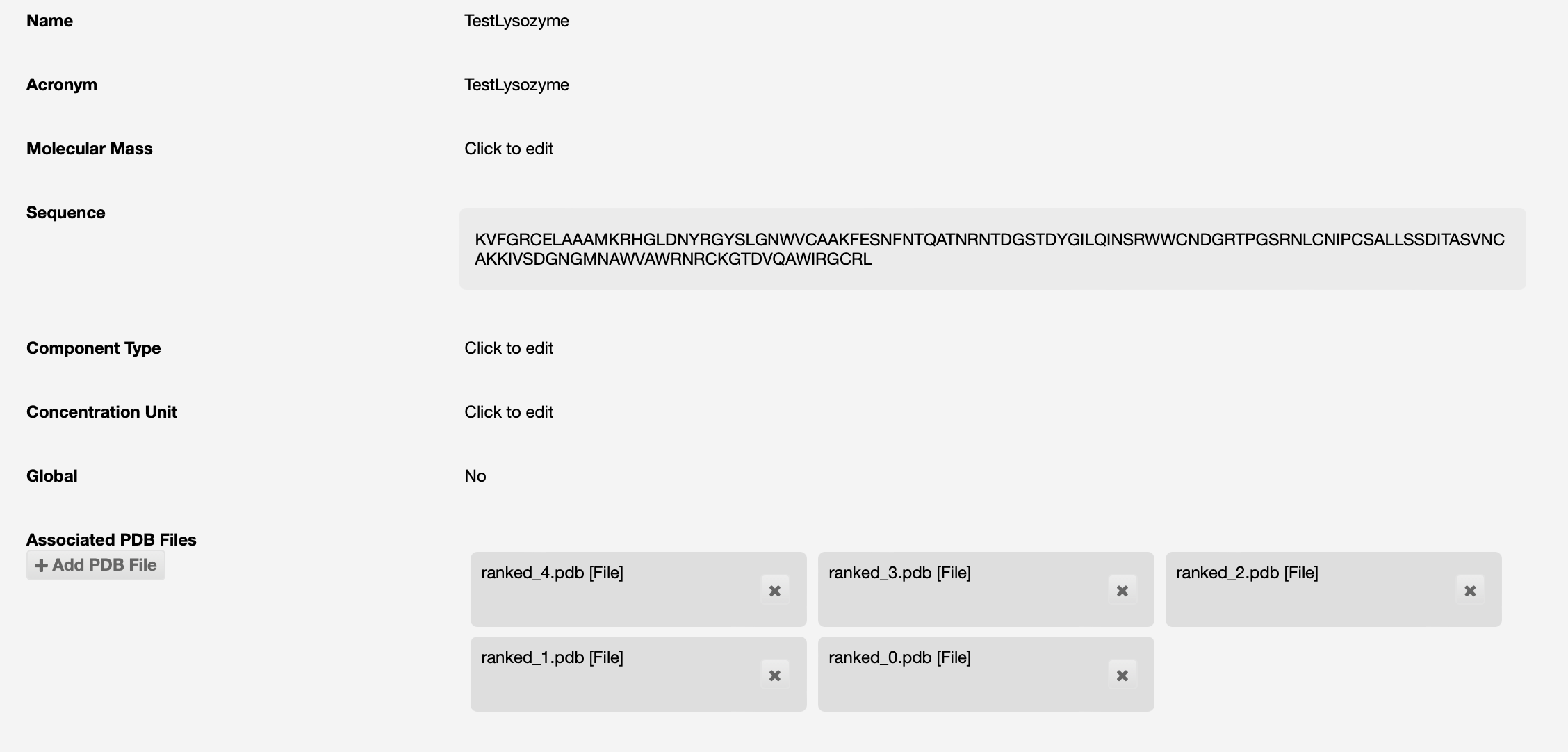
If you have a protein model that you expect to be a good source for molecular replacement, this should also be uploaded into ISPyB. These could be from the AlphaFold/EBI database, a RoseTTA fold model, or your own PDB structures.
After collecting your data the downstream processing can then be seen in DIMPLE and MrBUMP runs as seen on the collection window.
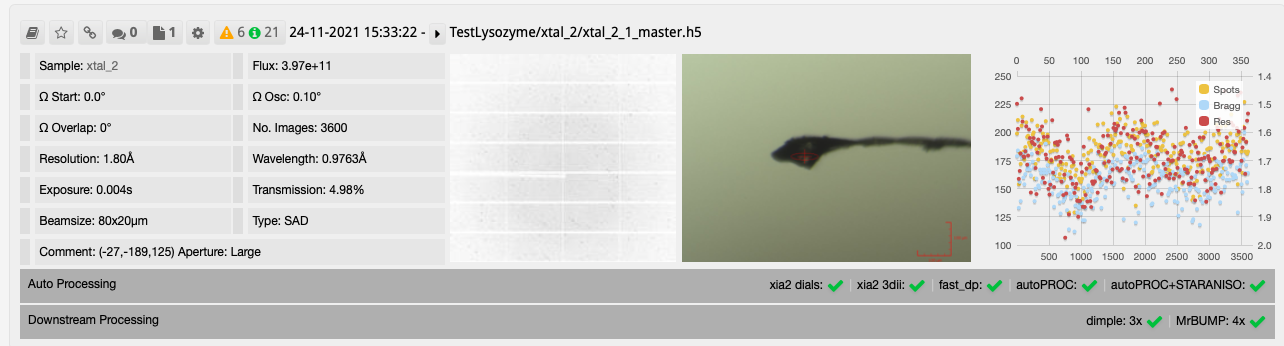
The results from DIMPLE using PDB models in ISPyB:
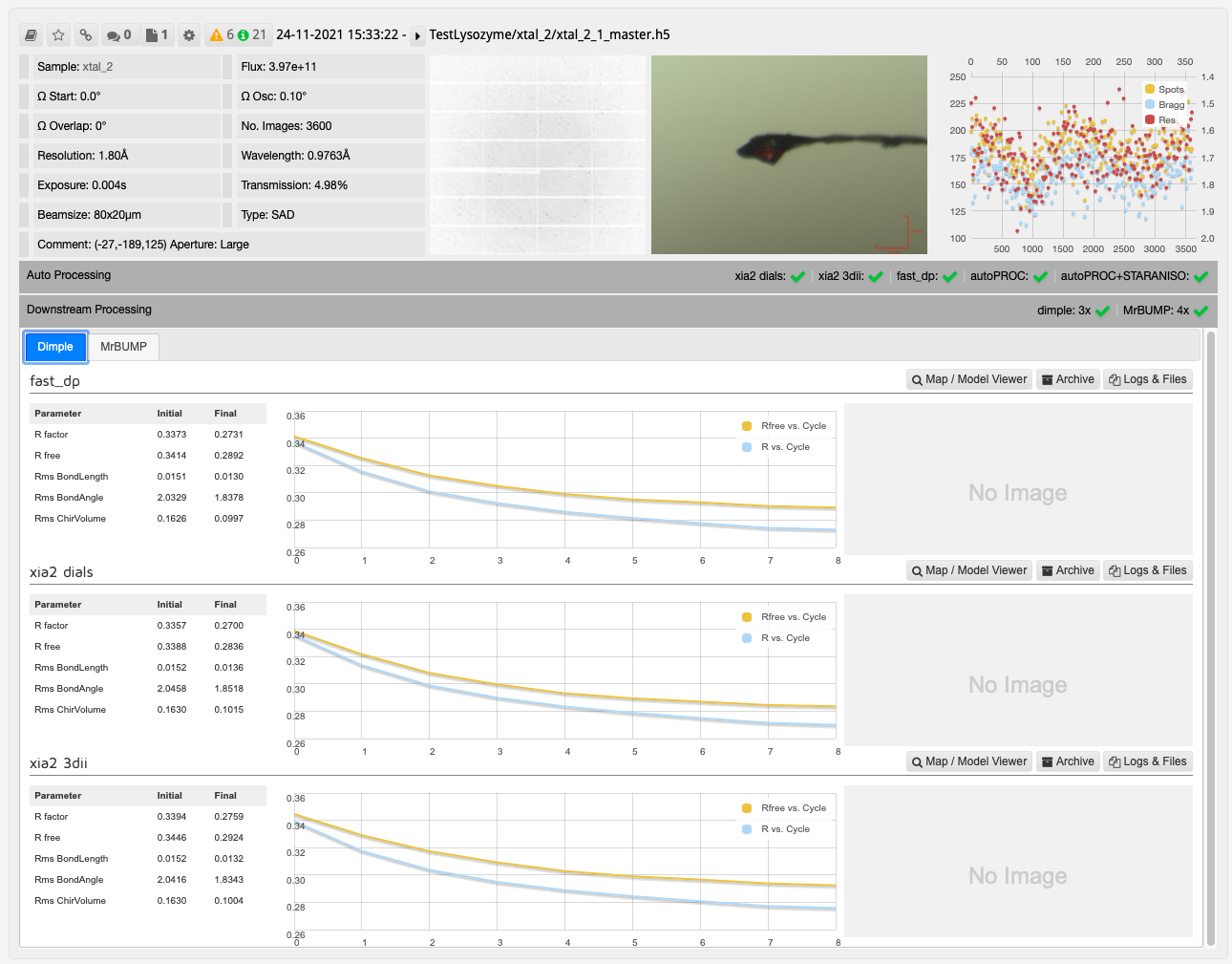
In this case, one run of MrBUMP with data processed with DIALS uses PDB files sourced from the protein databank following a sequence homology search, and the other uses any user provided PDB files and the AlphaFold models generated from the provided sequence. From here you can decide which has given the best overall results:
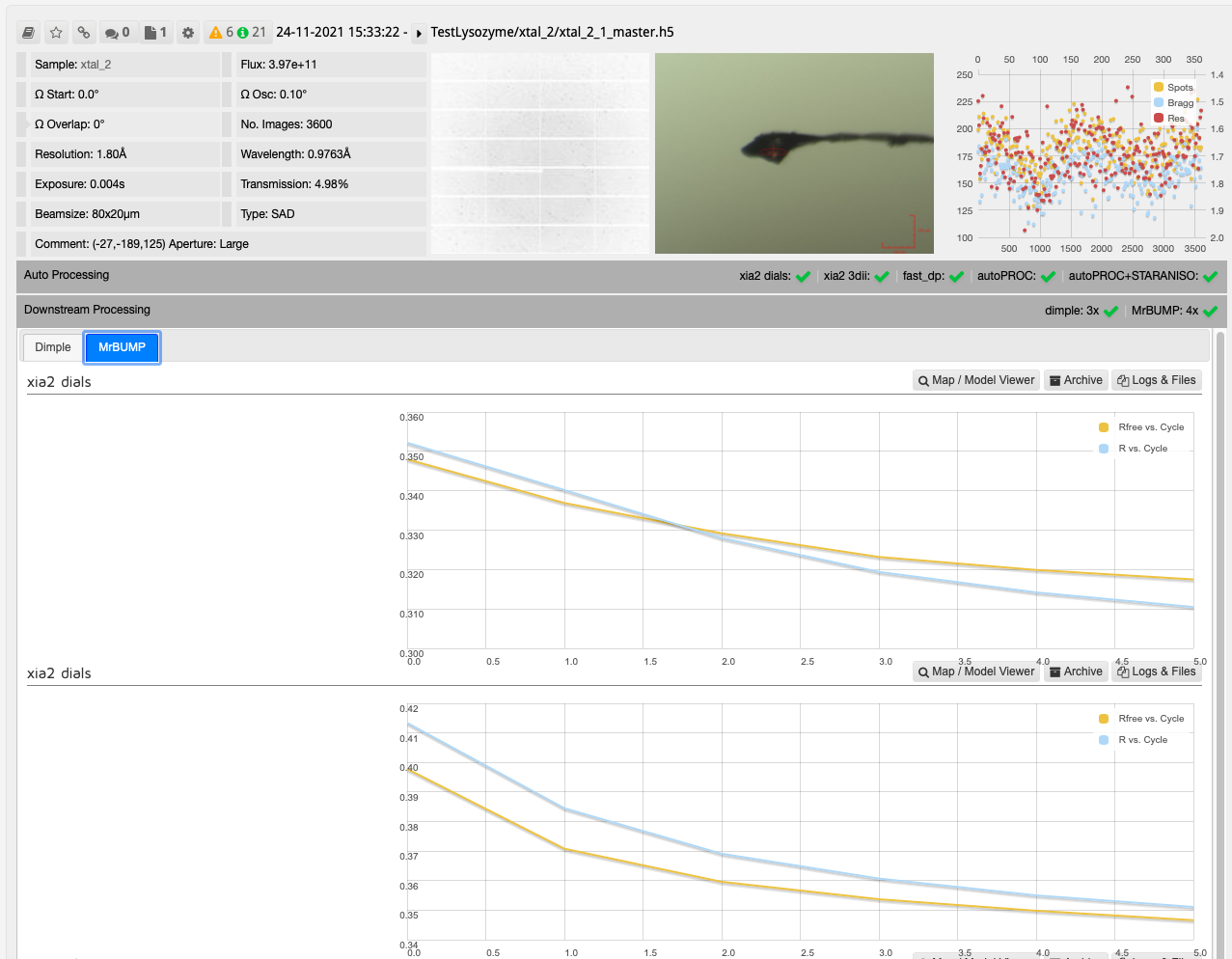
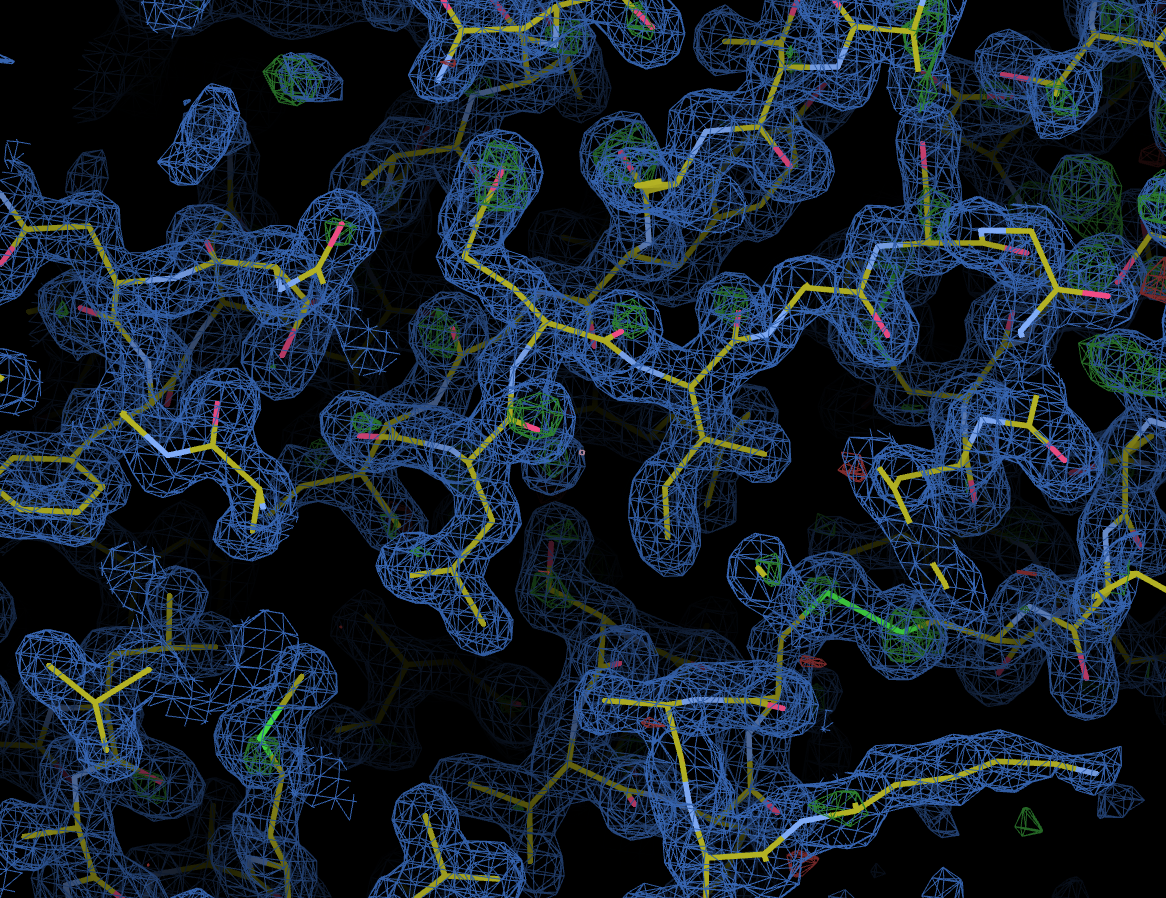
As with other downstream processing, models and maps can be viewed in UglyMol and results downloaded using the Logs & Files button:
Citations
Also remember to cite the appropriate publication for the autoprocessing pipeline used prior to downstream processing and the programs which are called by MrBUMP or DIMPLE,
Alphafold
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A. and Bridgland, A., 2021. Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), pp.583-589.
Alphafold EBI database
Keegan, R. M., & Winn, M. D. (2007). Automated search-model discovery and preparation for structure solution by molecular replacement. Acta Crystallographica Section D: Biological Crystallography, 63(4), 447-457.
DIMPLE
Wojdyr, M., Keegan, R., Winter, G., Ashton, A. (2013) DIMPLE - a pipeline for the rapid generation of difference maps from protein crystals with putatively bound ligands. Acta Cryst. A69: s299
RoseTTAFold

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.