In macromolecular X-ray crystallography, the avail ability of strongly diffracting crystals remains one of the major hurdles for successful structure determination. This is because projects studying membrane proteins, and large multi-component complexes with high solvent contents and weak packing of the molecules, are becoming more frequent. The diffraction quality of these crystals is often not sufficient to solve the structures.
ability of strongly diffracting crystals remains one of the major hurdles for successful structure determination. This is because projects studying membrane proteins, and large multi-component complexes with high solvent contents and weak packing of the molecules, are becoming more frequent. The diffraction quality of these crystals is often not sufficient to solve the structures.
Pre-crystallisation techniques:
 There is a number of pre-crystallisation techniques directed at improving the diffraction quality of macromolecular crystals. Some people try additives, seeding and alternative crystallisation for example. Others have to resort to more drastic aproaches such as reengineered constructs, alternative purification methods or even cloning related homologues. These techniques can be costly both in time and resources, and they assume that grown crystals are unchangeable and their diffraction cannot be improved.
There is a number of pre-crystallisation techniques directed at improving the diffraction quality of macromolecular crystals. Some people try additives, seeding and alternative crystallisation for example. Others have to resort to more drastic aproaches such as reengineered constructs, alternative purification methods or even cloning related homologues. These techniques can be costly both in time and resources, and they assume that grown crystals are unchangeable and their diffraction cannot be improved.
Post-crystallisation techniques:
 Post-crystallisation techniques (Heras & Martin, 2005, Newman, 2006) take into consideration the dynamic nature of macromolecular crystals. Proteins usually remain active and are normally capable of short and long-range reorganisation within the lattice (Morozov & Morozova, 1981, Morozova & Morozov, 1982, Morozov & Morozova, 1983, Kachalova et al., 1991). This reorganisation is often detrimental to the diffraction qualities of the crystal. However, in certain cases, reorganisation can lead to greater internal order, lower solvent content or a change in space group, that may result in improved diffraction or a higher quality dataset. Further techniques include soaking the crystal with different compounds, chemical cross-linking, crystal annealing and dehydration among many others (Abergel, 2004, Juers & Matthews, 2004, Harp et al., 1998, Heras & Martin, 2005, Newman, 2006, Yeh & Hol, 1998). In some cases, a combination of the above has proved necessary and successful (Nakamura et al., 2007).
Post-crystallisation techniques (Heras & Martin, 2005, Newman, 2006) take into consideration the dynamic nature of macromolecular crystals. Proteins usually remain active and are normally capable of short and long-range reorganisation within the lattice (Morozov & Morozova, 1981, Morozova & Morozov, 1982, Morozov & Morozova, 1983, Kachalova et al., 1991). This reorganisation is often detrimental to the diffraction qualities of the crystal. However, in certain cases, reorganisation can lead to greater internal order, lower solvent content or a change in space group, that may result in improved diffraction or a higher quality dataset. Further techniques include soaking the crystal with different compounds, chemical cross-linking, crystal annealing and dehydration among many others (Abergel, 2004, Juers & Matthews, 2004, Harp et al., 1998, Heras & Martin, 2005, Newman, 2006, Yeh & Hol, 1998). In some cases, a combination of the above has proved necessary and successful (Nakamura et al., 2007).

There are several ways of implementing these techniques; however, despite having proven successful in numerous cases, they are rarely used and are generally left for desperate cases. Furthermore, a controlled and systematic study of their effects has not been carried out as, in most cases, once the diffraction quality is sufficient for structure solution, the subject is not pursued further.
Dehydration as a Post-crystallisation Technique:
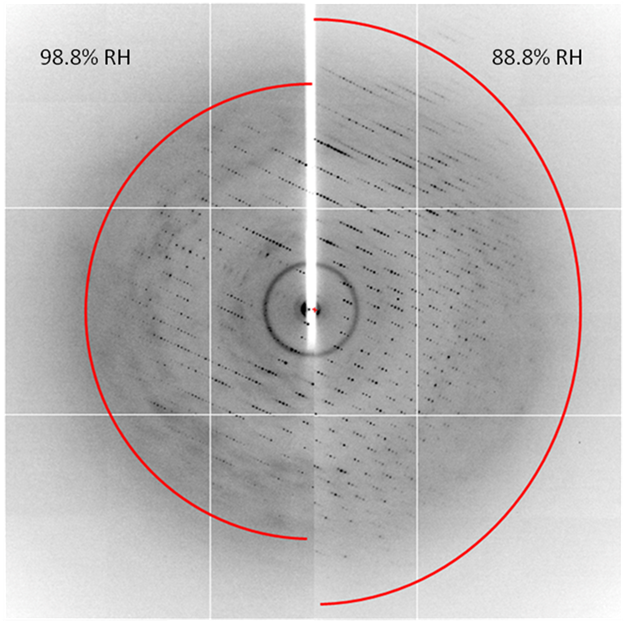 Dehydration is reported to be beneficial for the diffraction quality of macromolecular. In some cases the discovery has occurred as a result of a casual observation (cracked capillaries, drops left to dry, badly sealed trays, etc. (Abergel, 2004, Esnouf et al., 1998)). In others, it has been the result of a directed experiment to obtain the structure in a dehydrated state (Biswal & Vijayan, 2002, Kaushal et al., 2008, Saraswathi et al., 2002, Sukumar et al., 1999, Madhusudan et al., 1993, Kuo et al., 2003, Bowler et al., 2006a, Perutz, 1946, Amunts et al., 2007). Frequently, the effects of dehydration go unnoticed and are attributed to other factors such as crystal handling, new cryo-protectans or intrinsic variability in the crystals.
Dehydration is reported to be beneficial for the diffraction quality of macromolecular. In some cases the discovery has occurred as a result of a casual observation (cracked capillaries, drops left to dry, badly sealed trays, etc. (Abergel, 2004, Esnouf et al., 1998)). In others, it has been the result of a directed experiment to obtain the structure in a dehydrated state (Biswal & Vijayan, 2002, Kaushal et al., 2008, Saraswathi et al., 2002, Sukumar et al., 1999, Madhusudan et al., 1993, Kuo et al., 2003, Bowler et al., 2006a, Perutz, 1946, Amunts et al., 2007). Frequently, the effects of dehydration go unnoticed and are attributed to other factors such as crystal handling, new cryo-protectans or intrinsic variability in the crystals.
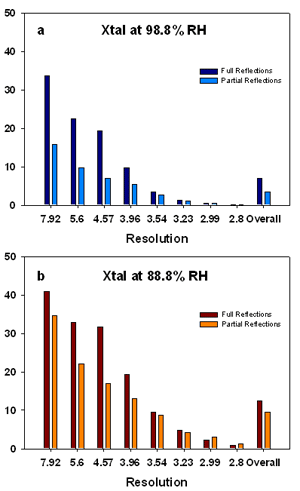 Several classic methods exist for protein crystal dehydration which include: simple air drying, vapour diffusion using salts, and soaking with dehydrating compounds (Heras & Martin, 2005, Newman, 2006). The advantages of these methods are that the experiment can be performed with very small humidity steps over long periods of time, and that a number of crystals can be modified at the same time. Unfortunately, the outcome of the dehydration is unknown until the experiment is finished and there is no direct way of assessing the progress of the experiment. This method is crystal- and time-consuming.
Several classic methods exist for protein crystal dehydration which include: simple air drying, vapour diffusion using salts, and soaking with dehydrating compounds (Heras & Martin, 2005, Newman, 2006). The advantages of these methods are that the experiment can be performed with very small humidity steps over long periods of time, and that a number of crystals can be modified at the same time. Unfortunately, the outcome of the dehydration is unknown until the experiment is finished and there is no direct way of assessing the progress of the experiment. This method is crystal- and time-consuming.
Most controlled dehydration studies have been carried out using vapour diffusion to regulate the rate of dehydration by using standard salt solutions (Greenspan, 1977). This method has been useful in a number of cases (Heras & Martin, 2005, Newman, 2006), including proteins of medical importance such as HIV reverse transcriptase (Ren et al., 1995, Stammers et al., 1994, Esnouf et al., 1998). The first attempts to create a purpose built dehydration chamber, and thus permit parallel data collection, were based on the use of CaCl2 to drive dehydration. Despite being efficient, they were difficult to control and reproduce. Nevertheless, this technique helped characterise numerous lattice changes in haemoglobin (Huxley & Kendrew, 1953) and a more ergonomic device was later developed (Pickford et al., 1993). Insulin also showed several dehydration transitions (Einstein & Low, 1962) and a modified version of the 1953 dehydration chamber was designed to work with it (Einstein, 1961). The modifications made to the device enabled thermo regulation above ambient temperature, making it possible to maintain a constant humidity state for several days (Einstein & Low, 1962). This work was the basis for the first gross molecular models of the protein (Einstein et al., 1963, McGavin et al., 1962).
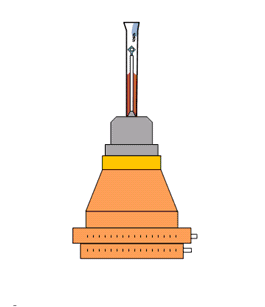 The latest addition to vapour diffusion methodologies has been the MicroRT system. This system, based on previous designs using glass capillaries and putty (Skrzypczak-Jankun, E. et al., 1996, Mac Sweeney, A & D'Arcy, A, 2003), uses a plastic capillary to surround the sample mounted on a modified copper pin (Kalinin & Thorne, 2005, Kalinin et al., 2005). Dehydration is achieved by changing the content of mother liquor placed at the tip of the capillary. This capillary system is simple and inexpensive, but retains the same practical issues of the older methods.
The latest addition to vapour diffusion methodologies has been the MicroRT system. This system, based on previous designs using glass capillaries and putty (Skrzypczak-Jankun, E. et al., 1996, Mac Sweeney, A & D'Arcy, A, 2003), uses a plastic capillary to surround the sample mounted on a modified copper pin (Kalinin & Thorne, 2005, Kalinin et al., 2005). Dehydration is achieved by changing the content of mother liquor placed at the tip of the capillary. This capillary system is simple and inexpensive, but retains the same practical issues of the older methods.
 Better suited to the sample environment is the wet vapour stream nozzle (Sjogren et al., 2002) designed to maintain a crystal at room temperature for spectroscopy studies (Sjogren & Hajdu, 2001a, b). This nozzle is much better suited to use at a synchrotron as no large modification of the beamline is needed to accommodate it within a standard environment, and beamline users do not need to change crystal mounting techniques. However, control of crystal humidity is difficult with this device.
Better suited to the sample environment is the wet vapour stream nozzle (Sjogren et al., 2002) designed to maintain a crystal at room temperature for spectroscopy studies (Sjogren & Hajdu, 2001a, b). This nozzle is much better suited to use at a synchrotron as no large modification of the beamline is needed to accommodate it within a standard environment, and beamline users do not need to change crystal mounting techniques. However, control of crystal humidity is difficult with this device.
 The lack of user friendly apparatus for dehydration experiments sparked an interest in designing a new device capable of generating a controlled humid airstream whilst allowing full functionality on most X-ray crystallography beamlines, in order for it to be widely and routinely used by the MX community. This is how the HC1 was born (Sanchez-Weatherby et al., 2009) . Since then it has been made available at the ESRF, MaxLab, Helmholtz-Zentrum Berlin (HZB), Diamond Light Source and the Canadian Light Source.
The lack of user friendly apparatus for dehydration experiments sparked an interest in designing a new device capable of generating a controlled humid airstream whilst allowing full functionality on most X-ray crystallography beamlines, in order for it to be widely and routinely used by the MX community. This is how the HC1 was born (Sanchez-Weatherby et al., 2009) . Since then it has been made available at the ESRF, MaxLab, Helmholtz-Zentrum Berlin (HZB), Diamond Light Source and the Canadian Light Source.

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.