Targeting a key COVID protein with antivirals
Jun 7, 2023
Jun 7, 2023
The SARS-CoV-2 virus that was responsible for the COVID-19 pandemic synthesises a large number of its proteins in a single polyprotein chain, and the virus relies heavily on a viral enzyme called the main protease to clip the polyproteins into individual units. The importance of the main protease makes it an appealing target for antivirals, but an in-depth understanding of the protease’s structure is required to determine how drugs may interfere with the enzyme. In a Nature Communications paper from March, Andre Schützer de Godoy at the University of São Paulo in Brazil and his colleagues collaborated with the Electron Bio-Imaging Centre (eBIC) at the Diamond Light Source to determine the structure of the main protease bound to its substrate by cryo-electron microscopy (cryoEM). This was one of the smallest protein structures to be solved at eBIC and led to important insights into how the active site engages with the polyproteins that it then cleaves into individual viral proteins which are essential for the viral replication. With their cryoEM structure in tow, the researchers explored how different antivirals interfere with the protease at the molecular level, including a drug developed by the COVID Moonshot Initiative in collaboration with Diamond. Their findings reveal that separate antivirals have forced the structure of the protease to change in distinct ways, paving the way to understanding how these drugs interfere with the enzyme at the molecular level.
SARS-CoV-2 and humans have distinct strategies for storing genes. The virus has an RNA genome, which is more prone to mutate through errors in replication than the DNA genome of humans. To reduce the risk of amassing mutations, RNA viruses keep their genomes small and compact. While the human genome has genes interspersed with non-coding sequences, viral RNA genomes fuse genes together end to end, partly as a strategy to economise on space. So-called polyproteins are synthesised from these fused genes and are subsequently cleaved into their individual viral proteins. SARS-CoV-2 encodes an enzyme called the main protease, which serves as the carving knife for polyproteins: It cleaves them into 12 smaller sections to free up the individual proteins starting with the protease itself. Scientists have begun designing drugs that target this protease so that they can block this critical carving process, a necessary step early during virus infection, and disrupt the virus from propagating within cells. This protease has already proven to be an effective drug target, with nirmatrelvir (brand name Paxlovid) approved for use in China, Europe, and the USA since 2022. Now the COVID Moonshot Initiative, a collaboration between Diamond and other institutes, seeks to develop other drugs that inhibit this protease. As such, researchers need to determine how both licensed and potential drugs interact with the protease at a molecular level, which in turn requires an understanding of how the protease matures into an active enzyme. Currently, it is known that multiple steps have to take place during protease maturation: the protease cuts itself out of the polyprotein at two positions, folds into a specific 3D shape, and pairs up to form dimers. However, it is still unclear in what order these events occur.
It was impossible to detect the running order of these events in previous studies as the protease matures too quickly. In hopes of tracking the steps during maturation, Andre Schützer de Godoy and his colleagues used a mutant form of the protease that differs by only one atom and matured at a far slower rate of up to two days. But when they used mass spectroscopy to observe all the states in which the protease exists, they detected every possible combination of clipped and non-clipped proteins, suggesting the cuts, folding, and dimerisation occur in no particular order.
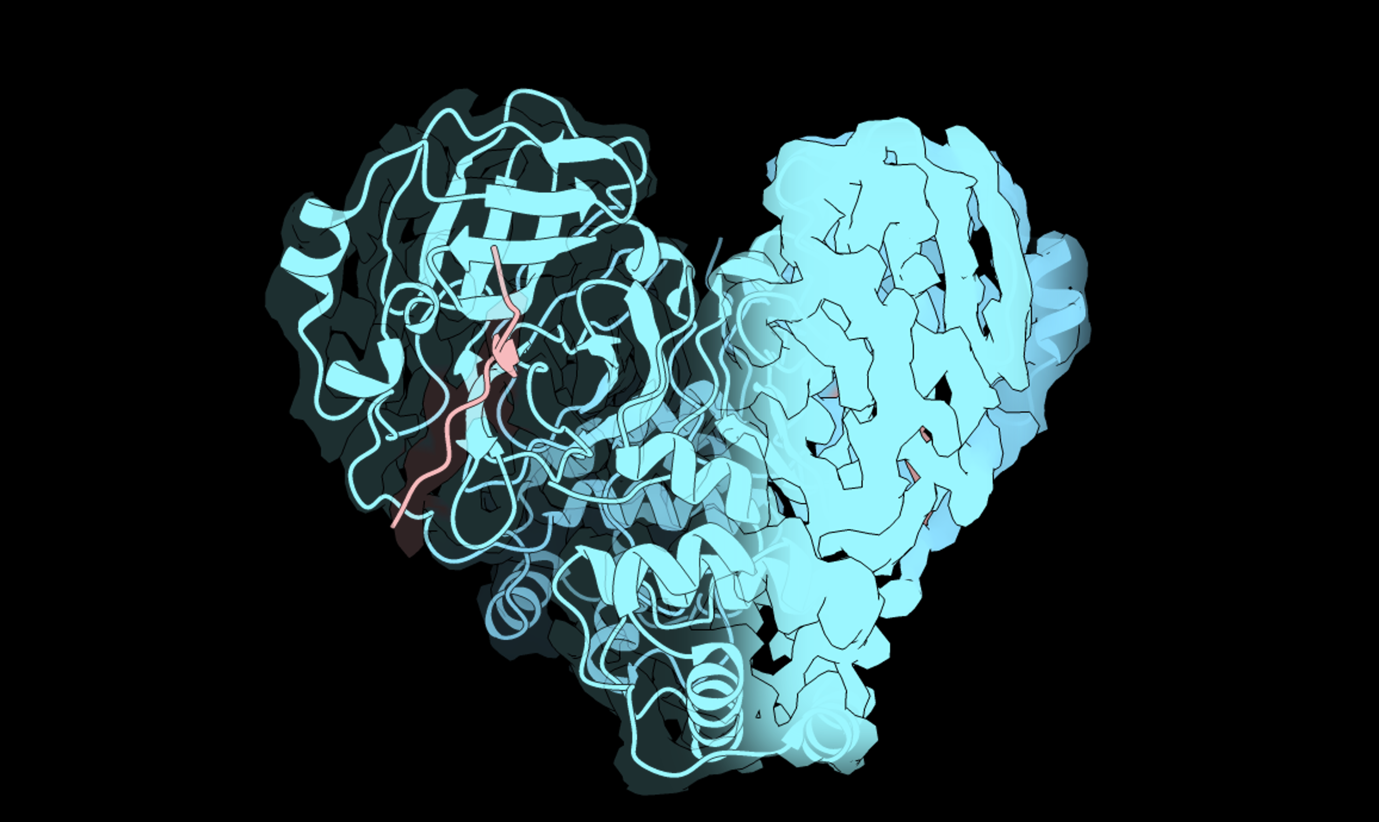
Having determined that the maturing protease exists in mixed states, the team sought to determine the structure of the complex. Although X-ray crystallography had been used to study this protease in the past, this technique struggles to resolve complexes with unfolded regions in multiple states. Cryo-electron microscopy was chosen as an alternative because different states could be separated and analysed individually with this approach. However, cryoEM posed new challenges because this technique has been optimised for protein complexes larger than the protease. Godoy says,
The rule of thumb is that larger particles like ribosomes are the easiest molecules to solve, and smaller proteins pose a greater challenge.
Using the state-of-the-art facilities at eBIC, the researchers successfully determined the structure of what is now the smallest protein to be resolved by cryoEM at Diamond. This dimer has a molecular weight of approximately 68 kiloDaltons, only 25 kiloDaltons larger than the current world-record for the smallest protein to have its structure solved with this technique. Godoy describes this work as a “landmark study” because although X-ray crystallography has been repeatedly used to study this protease, this is the only cryoEM structure available so far. By averaging together over 200,000 copies of the protein, they generated a refined structure of the protease dimer with a detailed view of its active site. What’s more, they found a protein fragment within the active site in multiple orientations, indicating that they had captured the protease processing its substrate. This allowed them to study how the active site contracts to cut the polyprotein and expands to release the divided products. Understanding the nature of this enzyme activity allowed the team to subsequently decipher the mechanisms by which antiviral drugs block the protease.
Dimerisation of the protease is required for its activity, and the researchers set out to determine how drugs affect this process. They used a technique called size exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) that can measure the size of protease particles to determine whether they were formed from individual copies or were paired-up into dimers. Nirmatrelvir appeared to encourage dimerisation. By capturing how the protease interacts with its substrate using cryoEM, the researchers could model how the drug interacts with the enzyme, revealing that dimerization was triggered by covalent bonding of nirmatrelvir to the active site. Future work is needed to determine if this blockade prevents the protease from processing polyproteins, but this brings the researchers a step closer to understanding how nirmatrelvir staves off the virus. The effects of a second inhibitory compound were tested as well, a drug lead developed by the COVID Moonshot Initiative that Diamond is part of. This compound exerted the opposite effect on the protease: it inhibited dimerisation and forced pre-existing dimers to separate, which indicates the drug prevents the protease from assuming its active state. Godoy cautions that it’s too early to tell if one of these compounds has more clinical potential than the other and more experiments are needed. Moving forward, his team wants to determine the structure of the protease in its singular monomeric state. Godoy says,
This has been a technical challenge because the particles readily form dimers. Monomers may be too small to resolve with cryoEM however.
Thus, his team are considering other techniques. By using X-ray crystallography at beamline I04-1, Godoy plans to investigate how the COVID Moonshot compound lead interferes with the monomer and prevents them from pairing into dimers. This structural work carried out at Diamond is integral to understanding how drugs interfere with viral proteins at a molecular level.
The study explored how the main protease in SARS-CoV-2 matures and dimerises into an active state. The structure of this protein was the smallest to be resolved at eBIC, and it revealed how the active site cleaves viral polyproteins. Drugs targeting this enzyme were found to inhibit it in different ways, including a lead compound for a new drug being developed by the COVID Moonshot Initiative, which Diamond is part of, which was observed to prevent the protease from pairing up into active dimers.
To find out more about eBIC or discuss potential applications, please contact Principal Electron Microscopist Daniel Clare: [email protected]
Noske GD, Song Y, Fernandes RS, Chalk R, Elmassoudi H, Koekemoer L, Owen CD, El-Baba TJ, Robinson CV, The COVID Moonshot Consortium, Oliva G, Godoy AS. An in-solution snapshot of SARS-COV-2 main protease maturation process and inhibition. Nature Communications 14(1545), doi: 10.1038/s41467-023-37035-5 (2023).
EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking (EUbOPEN grant n° 875510, RC).
NIAID of the National Institute of Health under award number U19AI171399.
Medical Research Council (MRC) program grant (MR/V028839/1).
Wellcome Trust, Grant No. 221795/Z/20/Z.
University of Oxford COVID-19 Research Response fund and its donors (BRD00230).
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES – Project 88887.516153/2020-00, ASG)
Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP projects 2013/07600-3, 2015/ 16811-3 and 2016/19712-9, GO).
TJE is a Junior Research Fellow at Linacre College and was supported by a Royal Society Newton International Fellowship.

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.