The mystery of allostery
Oct 6, 2017
Oct 6, 2017
A new allosteric drug screening approach has been developed by scientists hailing from The Francis Crick Institute in London and the University of Manchester. The revolutionary method, detailed recently in Nature Communications, yielded a surprising allosteric activator of a critical enzyme involved in the growth of the bacteria responsible for tuberculosis. The interaction between the intriguing activator and its enzyme was characterised at Diamond Light Source.
The enzyme ATP-phosphoribosyltransferase (ATP-PRT) is responsible for catalysing the first step in the biosynthesis of histidine in Mycobacterium tuberculosis. This enzyme is essential for the pathological progress of the bacteria and is absent in humans, so it is an attractive drug target. ATP-PRT is controlled allosterically by histidine, but the structure-function relationship of this regulatory interaction was not fully understood.
To shed some light on ATP-PRT regulation mechanism, a team of scientists embarked on a huge investigation which involved the development of a novel allosteric screening method. This technique unexpectedly discovered an allosteric activator of ATP-PRT known as 3-(2-thienyl)-L-alanine (TIH). TIH displayed some surprising properties, mainly that it could target the allosteric binding site of ATP-PRT and cause an opposite regulatory effect. Macromolecular Crystallography (MX) at Diamond’s Microfocus MX beamline (I04) permitted a closer look at this interaction, and it was noted that TIH triggered the same conformational change in ART-PRT as exerted by histidine, despite activating not inhibiting the enzyme. These findings could be just the start of a new era of allosteric drug discovery.
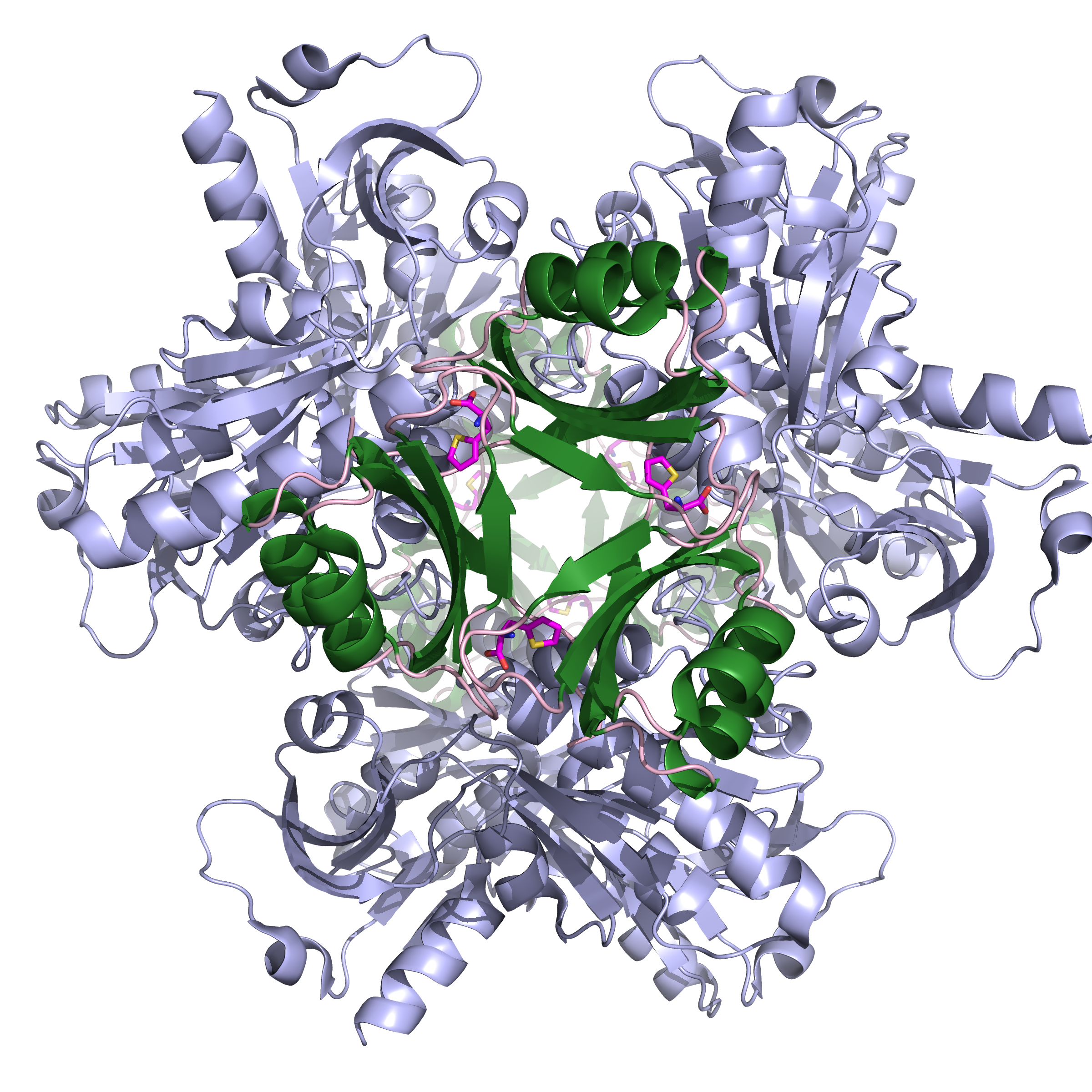
Figure 1: Ribbon representation of the close conformation of the homo-hexameric ATP-PRT bound to its small molecule activator (TIH). Catalytic domains are coloured in teal, allosteric domains in dark green and TIH is shown as ball and stick.
Biosynthesis of histidine
Tuberculosis is an infectious disease caused by the bacterium Mycobacterium tuberculosis, which largely affects the lungs and can often be fatal. Bacteria such as M. tuberculosis have the ability to make an amino acid called histidine, and they are reliant on this for their growth. The enzyme, ATP-PRT is crucial for the first stage in the biosynthesis of histidine, and as this enzyme is absent in humans it represents a favourable drug target for combating tuberculosis.
The activity of ATP-PRT is controlled allosterically, which means that a ligand (in this case histidine) affects its activity without binding to the active site. A team of scientists set out to understand this interaction and discover potential allosteric drugs against ATP-PRT. Senior group leader at The Francis Crick Institute and principal investigator of the study, Dr Luiz Pedro Carvalho explained their motivation: “There are occasions when drugs are made that are very potent and specific for the enzyme, but in the end are not effective when tested against bacterial cells. We believe that one of the causes of this is that enzymes should not be screened against in isolation. If you took ATP-PRT and ran a high-throughput screen against it and found an inhibitor, without knowing that the enzyme is always bound to some histidine, you might run into problems later on when you try to do things in vivo.”
Allosteric activator
Initially, the team devised a novel technique known as a Compound Screening in the Presence of an Inhibitor (CoSPI) assay to screen for allosteric regulators of ATP-PRT. CoSPI can identify molecules that only possess activity in the presence of a regulator-enzyme complex. In this case, the assay was conducted in the presence of histidine using 11 histidine analogues.
Dr Carvalho described the outcome of this approach: “What we found by running this assay was that instead of finding an inhibitor of the enzyme, we found an activator, called TIH. Allosteric activators are very rare so we wanted to characterise it.”
Unprecedented
Extensive kinetic analyses showed that the activator preferred to bind ATP-PRT in the presence of the inhibitor, exploiting the availability of the six allosteric binding sites of the hexameric enzyme (1 site per molecule), which was a huge surprise to the team. Both the inhibitor and activator were bound on the enzyme at the same time, which is unprecedented.
More work demonstrated that the activator was superior to the inhibitor. In fact, the enzyme became over-activated with TIH, which was a very uncommon observation. Structural characterisation of the enzyme at the Macromolecular X-ray Crystallography (MX) beamline (I04) at Diamond confirmed that the activator was binding to an allosteric site on ATP-PRT. By combining what was seen at Diamond with native ion-mobility mass spectrometry at the University of Manchester carried out by the group of Professor Perdita Barran, the team saw that the activator elicited almost the exact same conformational change to the enzyme that the inhibitor caused.
“The structures we obtained are static pictures of an equilibrium state so what probably is happening is that the inhibitor stabilises the interaction while the activator promotes it but does not stabilise it,” hypothesised Dr Carvalho.
More ATP-PRT modulators
The team are now in the process of determining how the interaction happens in a dynamic sense. Furthermore, they intend to return to CoSPI to carry out another search for ATP-PRT modulators after discovering that TIH was not able to penetrate M. tuberculosis.
As well as initiating the first steps to discovering a new drug against tuberculosis, the team have devised a new way of screening for potential enzyme modulators, so the researchers will continue to prove the worth of their novel approach.
To find out more about the I04 beamline, or to discuss potential applications, please contact Principal Beamline Scientist Dr Dave Hall: [email protected]
Pisco JP et al. Uncoupling conformational states from activity in an allosteric enzyme. Nat Comm 8, 203 (2017). DOI: 10.1038/s41467-017-00224-0.

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.