Annual Review: MX
Aug 3, 2016
Aug 3, 2016
 | ||
Insights into Parkinson’s disease from crystal structures of Parkin |
Related publication:
Wauer T, Simicek M, Schubert A, Komander D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370-374, doi:10.1038/nature14879 (2015).
Keywords:
Ubiquitin; Parkin; Parkinson’s disease; Enzyme regulation; E3 ligase; PINK1.
Parkinson’s disease affects millions of people worldwide, with the sporadic form typically occurring in people aged over 60 years. However, inherited versions of the disease can emerge in individuals who are in their twenties. Early-onset Parkinson’s disease is genetically linked to the proteins Parkin and PINK1, which perform crucial quality-control functions in the cell, and cause the condition when mutated.
Parkin is an E3 ubiquitin ligase, which is an enzyme that tags other proteins with ubiquitin to flag them for cellular processing. This high-level of control is aided by the activity of a further enzyme, the protein kinase PINK1, which adds a phosphate group to ubiquitin and activates Parkin. Prior studies showed that Parkin is trapped in an inhibited state and binding to phosphorylated ubiquitin was needed for activation, but the exact mechanism remained unclear.
By using Diamond’s Macromolecular Crystallography (MX) beamlines (I04 and I04-1), it was demonstrated how Parkin formed a complex with phospho-ubiquitin, which induced a conformational change. The shift in conformation released an inhibitory ubiquitin-like domain in Parkin from its position to drive the protein towards an activated state. The structure gave unprecedented insights in how Parkin works, and for the first time it was possible to delineate a sequence of events in PINK1-mediated Parkin activation, which were verified by biochemical and cell biological studies. This work could provide an essential starting point for the development of a causative treatment of Parkinson’s disease.Parkinson’s disease is the second most prevalent neurodegenerative disease in men and is associated with severe impairment in the quality of life and an increased mortality rate. The disease is characterised by the loss of dopaminergic neurons, but with only incomplete understanding of its molecular causes, no cure is currently available1. A powerful approach to study the underlying mechanisms of Parkinson’s disease are genetic factors that are linked with a disease outbreak at an early age. The most common cause for this genetic form of early-onset Parkinson’s disease are mutations in a protein called Parkin. Parkin is a member of the recently described class of RBR E3 ubiquitin ligases that mediate posttranslational modification of substrates with the small protein ubiquitin. As part of the cellular quality control Parkin attaches ubiquitin to membrane proteins in damaged mitochondria, which then triggers their degradation via mitophagy. Mutations in Parkin impair this process and the accumulation of damaged mitochondria lead to neuronal cell death, causing Parkinson’s disease1.
A first glimpse into how Parkin mediates ubiquitination was provided in 2013, when several groups including ours determined the first structures of Parkin, in our case with data collected at beamline I042. The structure not only gave insights in how the uncharacterised family of RBR E3 ligases works, but also allowed to map dozens of Parkinson’s disease patient mutations and classify them according to their pathogenic mechanism. It also revealed a ‘closed’ domain arrangement (Figs. 1a and 2) that locks Parkin in an autoinhibited state, suggesting that large scale conformational changes occur to activate Parkin. Surprising discoveries by us and others in 2014 showed that this activator is phosphorylated ubiquitin (phospho-ubiquitin), which is generated by the protein kinase PINK1, another Parkinson’s disease susceptibility gene3 (Fig. 1b). This work explained how PINK1 acts upstream of Parkin in mitophagy by recruiting and allosterically activating Parkin1. Structural and functional studies, with data collected at MX beamline I03, showed how ubiquitin phosphorylation changes its structure and affects many processes in the ubiquitin system4. However how phospho-ubiquitin activated Parkin remained elusive.
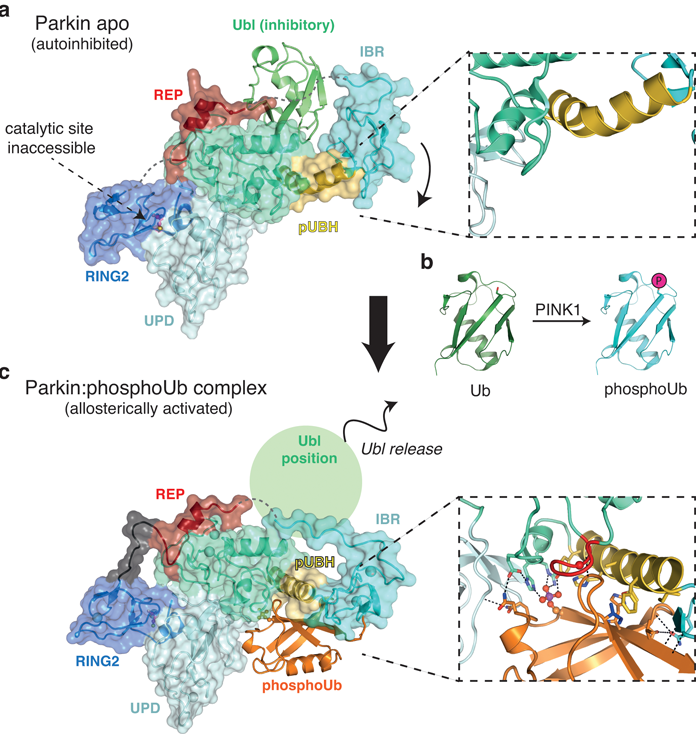
Figure 1: Conformational changes upon phosphoubiquitin binding. a) Apo structure of Parkin (pdbid: 4k955) reveals an autoinhibited conformation. The catalytic site is inaccessible and the inhibitory Ubl domain blocks E2 binding. The pUBH is kinked (box); b) Schematic generation of phosho-ubiquitin by PINK1, which is an allosteric activator of Parkin; c) Structure of Parkin in complex with phosphoubiquitin. Phospho-ubiquitin binding leads to IBR repositioning and subsequent release of the inhibitory Ubl domain. Highly specific interactions comprise the phospho-ubiquitin binding pocket and the pUBH is straightened out (box).
To address this question, the structure of Parkin in complex with phospho-ubiquitin was determined on beamline I04 (Fig. 1c). This revealed an unexpected binding site for phospho-ubiquitin, which is specifically held in place by amino acids coordinating the phosphate group and hydrophobic residues that interact with the isoleucine 44 patch, a common binding motif on ubiquitin (Fig. 1c). Mutations within the binding pocket fail to activate Parkin and do not recruit to mitochondria in cells. It also harbors previously unexplained patient mutations, confirming the importance of this activation event.
Intriguingly, phospho-ubiquitin initiates a dramatic conformational change in Parkin (Fig. 1). In the Parkin apo-structure, the IBR domain constitutes a platform for the inhibitory Ubl domain (Fig. 1a and 2 left). In this configuration the phospho-ubiquitin binding helix (pUBH; yellow in Fig. 1a) is kinked and allows for the IBR to reside in proximity to the Ubl. However, the specific interactions between phospho-ubiquitin and Parkin lead to a straightening of the pUBH upon phospho-ubiquitin binding (Fig. 1c). This pulls the IBR domain away from its Ubl stabilising location and repositions it adjacent to phosphoubiquitin, where it forms intermolecular interactions with the phosphoubiquitin C-terminus (Fig. 1c box). This conformational change hence releases the Ubl domain from its inhibitory position, and together with destabilising other autoinhibitory elements within Parkin, drive Parkin towards an activated state (Fig. 2b middle).
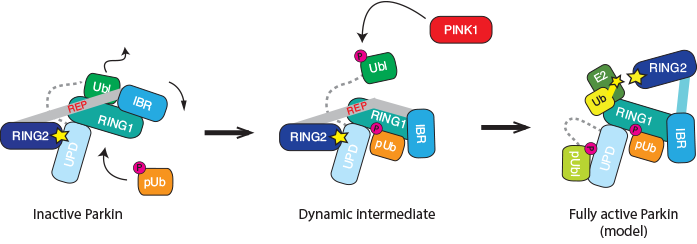
Figure 2: Cartoon representation of the Parkin activation path. Apo-Parkin is trapped in an autoinhibited conformation in which the active site is inaccessible (left). The allosteric activator phospho-ubiquitin triggers a conformational change and releases the inhibitory Ubl domain. This shifts Parkin towards a more active state. In addition, Ubl release allows for its phosphorylation by PINK1 (middle). Our biochemical data suggest that the phosphorylated Ubl domain binds back to Parkin, and stabilises the fully active, ‘open’ conformation. This requires structural validation.
PINK1 also phosphorylates Parkin in the Ubl domain, and this has been suggested as another mechanism to activate Parkin. However, the molecular details were less clear, in particular since elements crucial for the Ubl phosphorylation are blocked in apo structures of Parkin (Fig. 1a). As phosphoubiquitin binding leads to Ubl release, this could be a prerequisite for the efficient phosphorylation of Parkin, and biochemical experiments confirmed this. Indeed, our data indicated that the phosphorylated Ubl domain rebinds to the Parkin core via a second phosphate-binding site in the unique Parkin domain (UPD). This would open the Parkin structure completely and makes the catalytic site of Parkin accessible for full activation (Fig. 2 right). This model needs further experimental and structural verification.
Taken together, our work not only provides exciting insights on how Parkin is activated and mediates its crucial function in the cell, but is also a good example for the fruitful interplay between cell biological and structural insights. Moreover our structure might prove to be a critical first step to develop a causative treatment for Parkinson’s disease. The activation of Parkin has been shown to have a neuroprotective effect on dopaminergic cells, counteracting the loss of dopaminergic neurons as it appears in Parkinson’s disease1. It was clear from our mutational studies that non-physiological Parkin activation would lead to the autoubiquitination of Parkin and thus the likely impairment of the protein2. With the newly gained understanding of the cellular activation process, it might be possible to activate Parkin in a physiological manner that could counteract the effects of Parkinson’s disease. This could for example be achieved with a mimic of phospho-ubiquitin or a compound that releases the inhibitory Ubl domain. With the structural insights gained at the Diamond Light Source another step towards the development for Parkinson’s disease cure might have been made.
References:
Funding acknowledgement:
Work in the Komander lab is funded by the Medical Research Council [U105192732], the European Research Council [309756], and the Lister Institute for Preventive Medicine.
Corresponding authors:
Dr David Komander, MRC Laboratory of Molecular Biology, [email protected]; Tobias Wauer, MRC Laboratory of Molecular Biology, [email protected].

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.