Annual Review: Engineering & Environment
Jul 19, 2016
Jul 19, 2016
 | ||
Looking inside the radioactive box |
Related publication:
Stitt CA, Harker NJ, Hallam KR, Paraskevoulakos C, Banos A, Rennie S, Jowsey J, Scott TB. An Investigation on the Persistence of Uranium Hydride during Storage of Simulant Nuclear Waste Packages. Plos One 10, doi:10.1371/journal.pone.0132284 (2015).
Keywords:
Uranium; Hydrides; Corrosion; Oxidation; Tomography; Hydrogen.
A robust safety case is required for decommissioning nuclear waste at sites such as Sellafield. Substantial quantities of uranium are stored in a range of environments at different stages of corrosion. During corrosion, metal oxides and hydrogen gas are produced. As hydrogen accumulates, the corrosion of uranium may switch to produce uranium hydride (UH3) instead. This pyrophoric compound reacts vigorously with oxygen to make UO2, and as such is thought to exist only fleetingly.
Researchers wanted to investigate the risk posed by an accumulation of pyrophoric uranium hydride. For these experiments, UH3 was stored underwater after being artificially formed on a uranium rod and encased in the grout used at nuclear waste facilities. The sample underwent periodic testing at 0, 3, and 10 months on the Joint Engineering, Environmental, and Processing beamline (I12).
This beamline offers micron-scale X-ray tomography imaging of corrosion pits, and structural identification of corrosion products using X-ray powder diffraction. High energy synchrotron X-rays penetrate the grout casing and examine uranium corrosion underneath without breaking the confinement – crucial for the study of highly reactive air-sensitive UH3.
Results showed that UH3 not only persisted, but the sample volume increased by 4.5% over the last seven months as more, lower density corrosion products were generated. Grout acts as a physical barrier, limiting the access of reactive species to the uranium surface and so delaying oxidation.
Metallic uranium is strongly reactive in the presence of oxidising species. In water or water vapour, the recorded rates of uranium corrosion are observed to be notably faster than in air or dry oxygen and just as for aqueous corrosion of any other metal, the arising by-product of the water-uranium reaction is hydrogen1,2. If a sufficient concentration of hydrogen accumulates then the corrosion of uranium may switch from forming uranium oxide to forming uranium hydride (UH3). Uranium hydride is highly pyrophoric due to its vigorous and strongly exothermic reaction with oxygen (in air) to form UO23. This highly reactive behaviour has been attributed to a number of previously documented thermal excursions at nuclear waste facilities. As a consequence, the presence and persistence of UH3 in any nuclear waste storage scenario is regarded as highly undesirable because it presents an additional risk factor for the safe management of the waste.
The observed reactivity of UH3 has led to the assumption that it cannot persist for any significant period in a storage environment at ambient temperatures; wet or dry. If this assumption is correct, then the long term safety risk posed by hydride is significantly decreased as its existence is both transient and brief.
As the planned start for decommissioning draws closer, there is a requirement to provide an experimental verification of whether UH3, formed in a waste storage environment, can persist for any significant period. This was the focus of our work at Diamond on the I12 beamline.
A uranium coupon sourced from virgin fuel material was mechanically polished to a P2500 SiC grit finish and cut into a cuboid measuring 20 x 0.5 x 0.5 mm. This sample was then treated in 5M HNO3 for 3 hours to remove the surface oxide, then rinsed with water and cleaned for 5 minutes in an ultrasonic bath with ultrapure acetone and then methanol. After this procedure the sample had visibly tarnished, forming a thin oxide layer on the surface. This oxide was left to grow in air for a further 15 minutes to ensure that a complete coherent oxide had been formed across the entire sample. This ensured that all subsequent reactions with the metal had to occur through this interface. The sample was then encapsulated in grout using a 3:1 mixture of Blast Furnace Slag (BFS) to Ordinary Portland Cement (OPC) and prepared with a 0.4 water to cement ratio. It was then cured for 3 days in a moist atmosphere.
Further curing was allowed for a week in laboratory atmospheric conditions. Subsequently, the sample was degassed under vacuum (5 x 10-9 bar) in a gas rig for 8 hours at 80 °C and then heated for a further 16 hours at 170 °C to dry the grout before being exposed to a fixed volume of H2 at 0.6 bar. These conditions were chosen to give the best chances of forming a pure β-UH3. After 0.012 mmol H2 uptake was recorded (which corresponds to approximately 3.5% corrosion of the uranium sample to form UH3), the reaction was halted by evacuating all H2 from the system and cooled under vacuum. The sample was then analysed on I12 in its initial state and then after 3 months in 40 ml of de-ionised water, and then after a further 7 months in the same de-ionised water bath. In each case the sample was dewatered at 60 °C overnight under vacuum to remove excess surface and pore water, in order to conform to safety restrictions.
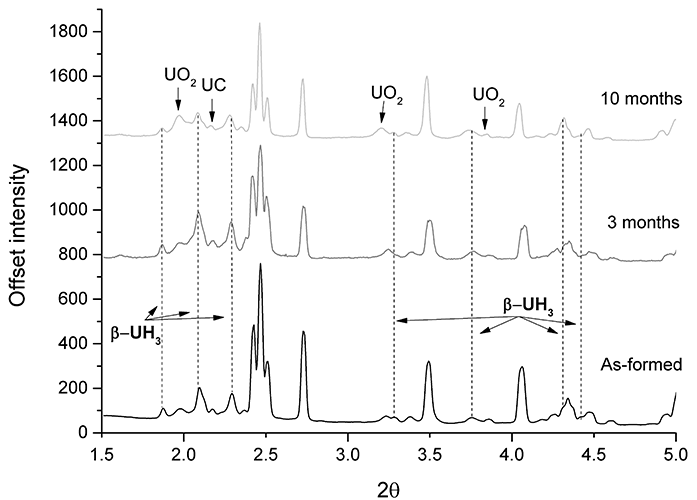
Figure 1: XRPD results of hydrided uranium, encapsulated in cement after 0, 3, and 10 month submersion in de-ionised water.
X-ray tomography (XRT) was used to image the sample corrosion, whilst X-ray Powder diffraction (XRPD) identified the UH3 corrosion products at each consecutive stage. The X-ray energy for XRPD was chosen around 114 keV.
XRPD results of the three sample examination periods are displayed in Fig. 1, clearly indicating that β-UH3 is present on the surface of grout-encapsulated uranium. A peak ascribed to UC shows the high impurity content of Magnox uranium. The data was normalised to the first UO2 peak, and all unlabelled peaks were ascribed to uranium.
Figure 2 displays the 3D tomographic reconstructions of the uranium sample (top) and corrosion layer (bottom) before and after 3 and 10 months submersion in water, the grout is omitted.
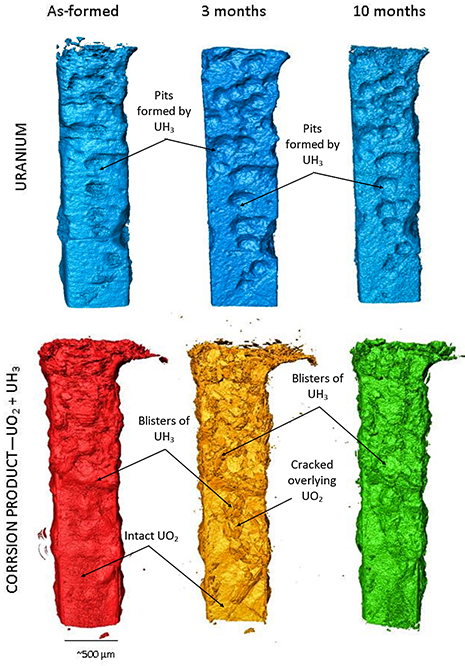
Figure 2: Three dimensional representations of the uranium samples encapsulated in cement at each stage of the 10 month period. Top shows the uranium volume, bottom shows the corrosion volume.
On first inspection, two morphologies of corrosion products were identified on the sample at each stage: i) deep pits in the metal filled with blisters of less dense corrosion product that was flaky and porous at the surface, characteristic of UH3 (Fig. 2 top); and ii) an enveloping layer blanketing the remaining surface of the metal attributed to surface oxide; UO2 (Fig. 2 bottom). Over the 10 month period submersed in water, no significant changes in the overall corrosion layer could be identified; however, measurements of the corrosion layer suggest a volume increase of 4.5% average ±3% between analysis at 3 and 10 months. A volume increase would be expected from the corrosion of uranium to either UO2 or UH3, as these corrosion products are almost half the density of the metal.
Figure 3 shows a sequence of tomography slices of the same position for each time period, and showing that despite a growing corrosion layer, some corrosion product identified as hydride on the basis of blister like morphology. Fractures parallel to the metal surface were also revealed within hydride blisters, highlighting the fragile nature of the material.
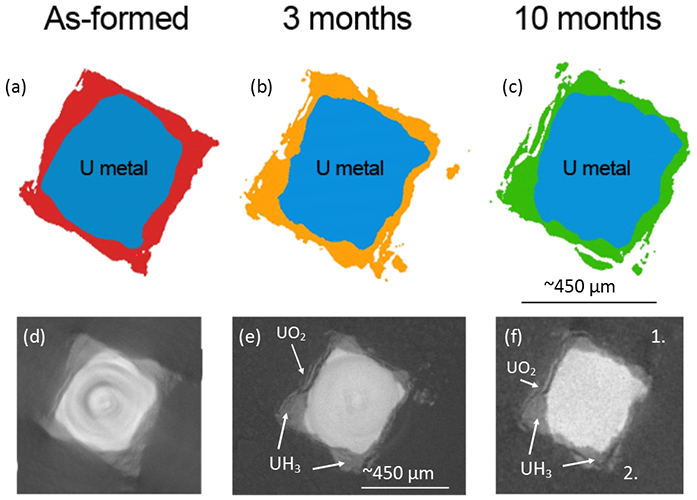
Figure 3: Cross sections at the same position of the uranium encapsulated in grout over time. Images (a) to (c) are accurate representations of the uranium (blue) and surrounding corrosion products (red, orange and green) perpendicular to the length of the sample. Images (d) to (f) are radiographs corresponding to the above representations, but with the additional surrounding grout.
The results obtained from this experiment provide strong evidence that β-hydride artificially formed on grout encapsulated uranium will persist during storage in water for a period of 10 months at room temperature. The results also confirm that under the conditions studied UH3 is less susceptible to rapid and continued oxidation (and perhaps pyrophoricity) in a grouted system than in air.
The present work has demonstrated that laboratory made β-UH3 will persist on grout encapsulated uranium submersed in a semi sealed volume of water for periods up to 10 months. X-ray diffraction data indicated β-UH3 and UO2 peaks were present after 3 and 10 months submersion in water. X-ray tomography allowed comparisons of uranium and uranium hydride oxidation rates to determine that the conditions within the grout were likely an anoxic H2O system. In addition, it was identified that oxidation was limited by the encasing grout body. This was identified as a legitimate cause for the current and future persistence of hydride in grout.
The next logical step for ongoing research is to understand the location and abundance of the UH3 and UO2 within the grout system. Neutron tomography or secondary ion mass spectrometry would provide further understanding of the UH3 oxidation rates in the current sample, which will be used to produce rates for larger wet storage systems.
References:
Funding acknowledgement:
This work is primarily supported by a jointly funded EPSRC-NDA grant (EP/I036397/1) entitled ‘In situ time-dependent characterisation of corrosion processes in nuclear waste storage and GDF environments’. The materials and additional financial support for this project has come from Sellafield Ltd. as part of a platform grant associated with the Centre of Expertise for Uranium and Reactive Metals. In the writing of this paper Sellafield and RWM Ltd have had input and provided permission to publish.
Corresponding author of this submission:
Dr Thomas Scott, University of Bristol, [email protected].

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.