The majority of bacterial gene regulators bind as symmetric dimers to palindromic DNA operators. Multimeric forms of proteins, including tetramers, are able to recognize longer operator sequences in a cooperative manner, although how this is achieved is not well understood due to the lack of complete structural information. Using data collected at Diamond Light Source beamline I04, we solved the crystal structures of the multidrug binding protein TtgV, a gene repressor that controls efflux pumps, on its own and in complex with a 42 bp DNA operator containing two TtgV recognition sites at 2.9 and 3.4 Å resolution respectively. These structures represent the first full length functional tetrameric protein in complex with its intact DNA operator containing two continuous recognition sites. TtgV binds to its DNA operator as a highly asymmetric tetramer and induces considerable distortions in the DNA resulting in a 60o bend. Furthermore, upon binding to its operator, TtgV undergoes large conformational changes at the monomeric, dimeric and tetrameric levels. The structures reveal a model for cooperative DNA binding of tetrameric gene regulators that are applicable to other tetrameric proteins and suggest a reinterpretation of previous structural model of tetrameric gene regulators in the literature.
Bacterial gene regulators are model systems to study protein-DNA and protein-ligand interactions as well as principles of gene regulation. Many specialized gene regulators, such as the CAP activator, bind to highly conserved DNA operator sequences with high affinity. However, some global regulators bind to a wider range of less conserved DNA sequences and more complex regulatory systems have evolved to ensure binding specificity. One strategy is to recognize a longer region of DNA using multiple, less conserved, recognition sites and with reduced specificity within each individual site. A large number of bacterial gene regulators adopt this strategy and use tetramers to recognize two DNA sites. Some of the best-studied examples include the Lac repressor (LacI or LacR) and the lambda repressor (?cI).1-3 Although this mode of recognition is widespread, there is no structural information on a tetrameric protein bound to a continuous DNA operator containing two or more binding sites, which has hindered our understanding of cooperative binding by tetrameric gene regulators and the mechanism of their activation. However, tetrameric models of protein/DNA complexes have been proposed based on partial domain structures and incomplete DNA sequences. One such example is the tetrameric LacI/DNA complex model derived from the 4.8 Å resolution structure of a dimeric LacI/DNA complex and a tetrameric structure of the C-terminal domain.1
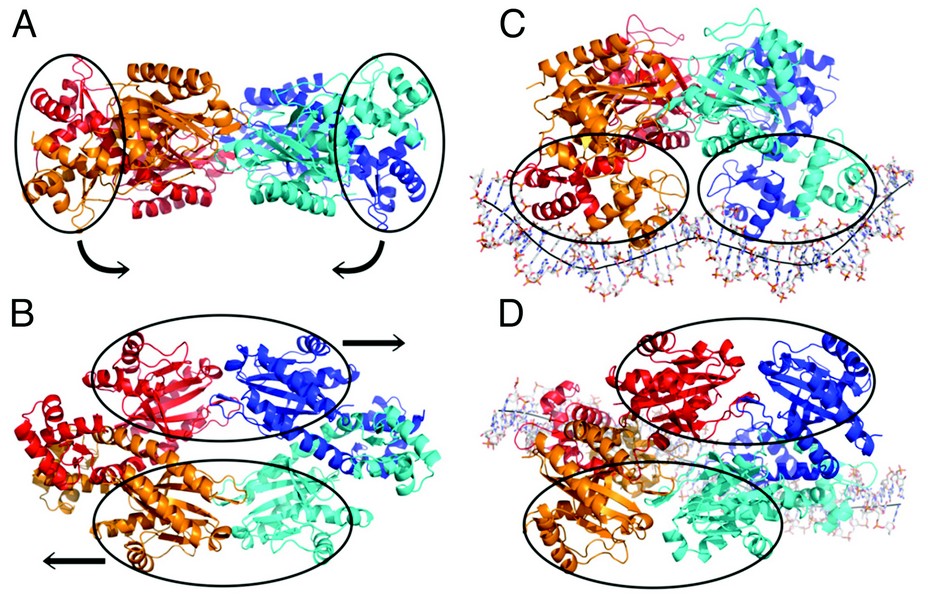
Figure 1: Crystal structure of TtgV alone viewing from the side (A) and the top (B) and in complex with its intact DNA operator viewing from the side (C) and the top (D).
We have solved the crystal structures of a full length TtgV tetramer and its complex with an intact 42 bp DNA operator that contains two TtgV recognition sites. The TtgV monomer consists of a N-terminal DNA-binding domain (DBD), a C-terminal ligand-binding domain (CTD) and a linker helix (Fig. 1, Fig. 2). In the TtgV alone structure, the monomers form a symmetric tetramer through their CTDs (Figs. 1A, 1B). In the DNA complex structure, the CTD tetramer lacks the symmetry observed in the TtgV alone structure. Instead it forms a skewed diamond shape (Figs. 1C, 1D). The large differences between the TtgV alone and TtgV/DNA structures arise from the differences at the monomeric, dimeric and tetrameric levels. At the monomeric level, the linker helix between the DBD and CTD forms a continuous helix with the first helix in CTD (a5) in the TtgV apo structure. However, in the TtgV/DNA complex, the protomers adopt two different conformations, the linker helix is bent relative to a5 in one protomer while the linker helix is partially unwound in another (Fig. 2). The consequences of these changes are reflected in the reduced stability of the tetramer assembly, with a total buried interface of ~9500 Å2 instead of ~11000 Å2 as in the symmetric apo structure.
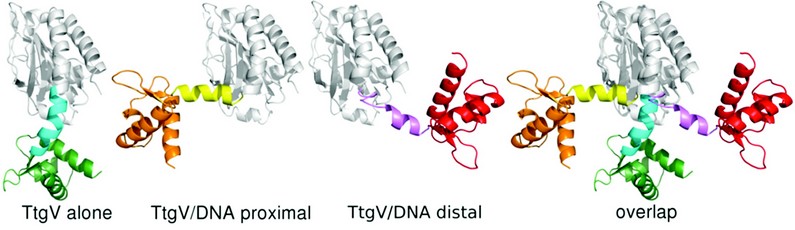
Figure 2: Two different TtgV monomer conformations were found in the TtgV/DNA complex compared to TtgV alone structure. There are dramatically different orientations of the linker helix (cyan, yellow and magenta) which result in different locations of the DNA-binding domain (orange, green and red) in relation to the C-terminal domain (white).
Cooperativity is observed widely in biological systems that involve protein oligomers. The general model for cooperativity in ligand binding proposes that two distinct functional states (Tense state (T-state) or Relaxed state (R-state)) exist. The T-state is more stable but incompetent in ligand binding. The R-state, on the other hand, is less stable but has higher affinity for the ligand. A concerted or a sequential model 4 has been used to describe cooperative binding. The concerted model has been generally applied to oligomeric proteins that have identical ligand binding sites. It argues that the protein exists in equilibrium of either of two states (T- or R-state) and all subunits within the oligomer exist in the same states, all in T- or R-state. The sequential model, on the other hand, assumes that ligand binding induces changes in one subunit (induced fit from T- to R-state) which could then influence ligand binding in other subunits.
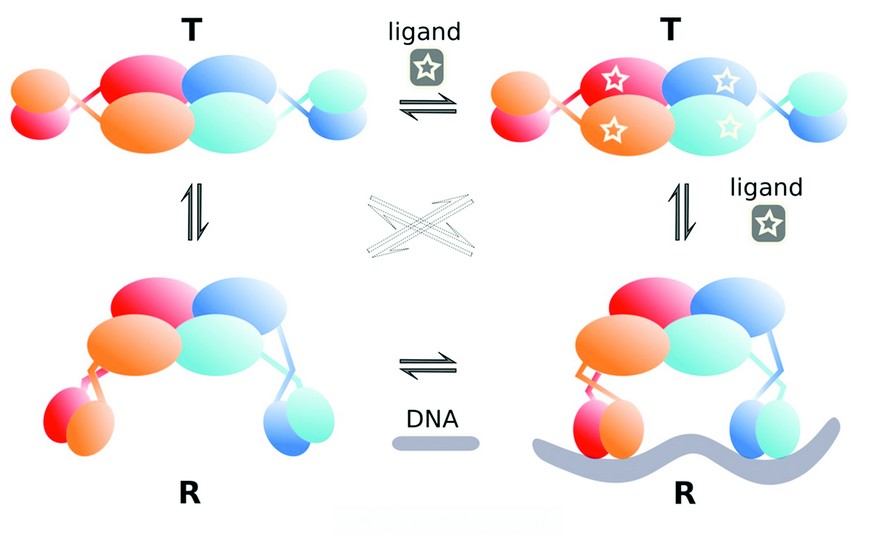
Figure 3: Proposed cooperative binding model for TtgV tetramer. In the absence of ligand or DNA, TtgV exists in the equilibrium of T and R states. Only R state can bind to DNA.
The two distinct conformations observed here reveal that a concerted cooperative binding model could be applied to the DNA (ligand) binding of tetrameric gene regulators with a dimer as a single subunit (as each dimer binds to one DNA site). The TtgV alone structure represents the T-state which is more stable but has lower affinity for the ligand (DNA). The DNA complex structure, on the other hand, represents the R-state which is less stable but has a higher affinity for the ligand (DNA). In the absence of DNA, the protein predominately exists in the more stable T-state. However, upon ligand binding, the favorable binding energy between the protein and the ligand can offset the higher energy cost of the R-state, switching the protein to the R-state (Fig. 3). Many tetrameric gene regulators show identical dimer conformations within the tetramer and we argue that two distinct conformational states exist (T and R) and the concerted cooperative binding model presented here is applicable irrespective of the exact structures and conformations.
Our structures also reveal large distortions imposed on the DNA. Overall, the central axis for the DNA double helix is W shaped, with inward bends at the narrowed minor grooves (~ 40o) that are A/T rich while a convex kink at the widened minor groove with a G/C pair in the middle of the operator, reducing the concave bend over the entire operator to 60o (Fig. 1C). The large distortions in the DNA are a direct consequence of the interactions between the TtgV tetramer and the DNA operator. Likewise, the asymmetric structure observed in the TtgV/DNA structure is due to the constraints of binding to two DNA sites. Our structures therefore suggest that the structural models of a tetramer/DNA assembly derived from partial structural information are unlikely to represent the true assembly of the gene regulators.
Lu, D., Fillet, S., Meng, C., Alguel, Y., Kloppsteck, P., Bergeron, J., Krell, T., Gallegos, M.T., Ramos, J., Zhang, X. Crystal Structure of TtgV in complex with its DNA operator reveals a general model for cooperative DNA binding of tetrameric gene regulators. Genes Dev. 24(22), 2556-65 (2010)
References
- Lewis, M. et al. Science. 271, 1247-1254 (1996).
- Stayrook, S. et al. Nature. 452, 1022-1025 (2008).
- Schumacher, M. A. et al. Embo J. 21, 1210-1218 (2002).
- Fersht, A. Structure and mechanism in protein science. W.H. Freeman and Company, New York (1999).
Funding Acknowledgement
Medical Research Council to XZ with earlier funding from Human Frontier Science Foundation and the Royal Society.
