Towards a structural understanding of transport within the proton dependent oligopeptide transporter (POT) family
Jun 2, 2011
Jun 2, 2011
The proton dependent oligopeptide transporters (POTs) are a large family of integral membrane proteins that use the inwardly directed proton electrochemical gradient to transport small peptides, amino acids and nitrate across cellular membranes in both pro- and eukaryotic cells. Evolutionarily the POT family sits within the much larger Major Facilitator Superfamily (MFS), members of which contain a common structural motif of 12 transmembrane-spanning alpha-helical segments. The human genome contains four members of this family, two of which, PepT1 and PepT2 are responsible for the absorption of dietary peptides in the small intestine and peptide re-absorption in the kidney. Peptide transporters also contribute significantly to the oral bioavailability and pharmacokinetic properties of a number of important drug families, such as the beta-lactam antibiotics. To gain further insight into the molecular mechanism of drug and peptide transport, we determined the crystal structure of a prokaryotic member of the POT family, PepTSo, with similar substrate specificity and a high degree of sequence conservation to the mammalian PepT proteins. The structure of PepTSo, together with our associated kinetic data, provides valuable new insights into mammalian peptide transport and provides the starting point for further structural and biochemical studies on this pharmaceutically important transporter family.
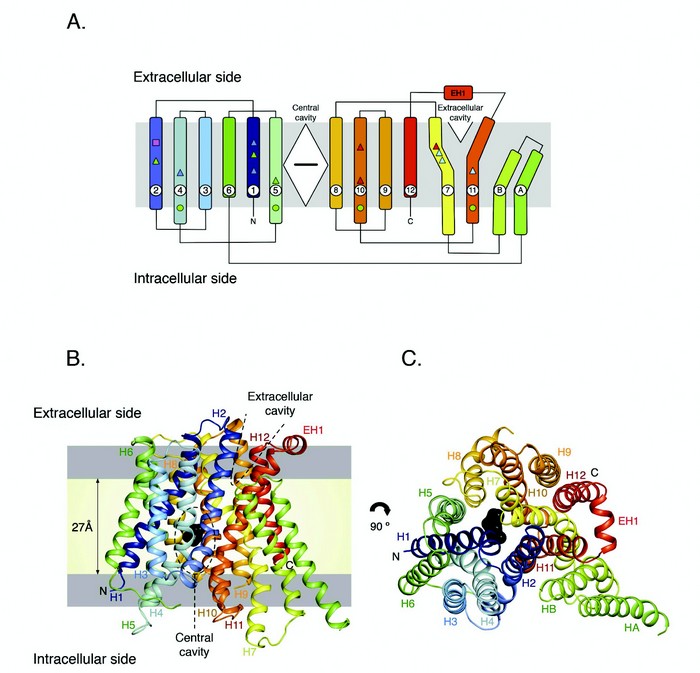
Figure 1: Structure of PepTSo. (A). PepTSo topology. The central and extracellular cavities are shown as a closed diamond and open triangle, respectively. A bound ligand in the central cavity is represented as a black horizontal bar. Functionally important residues conserved between PepTSo and metazoan peptide transporters are highlighted by shapes and mapped onto the topology diagram. (B). PepTSo structure viewed in the plane of the membrane. The two hydrophilic cavities present in the structure are outlined in dashed lines. The hydrophobic core of the membrane (pale yellow) is distinguished from the interfacial region (light grey). N and C represent the N- and C-termini respectively. Bound ligand is shown in black. Helices are labeled. (C). View from the extracellular side of the membrane. (Reproduced from publication with permission).
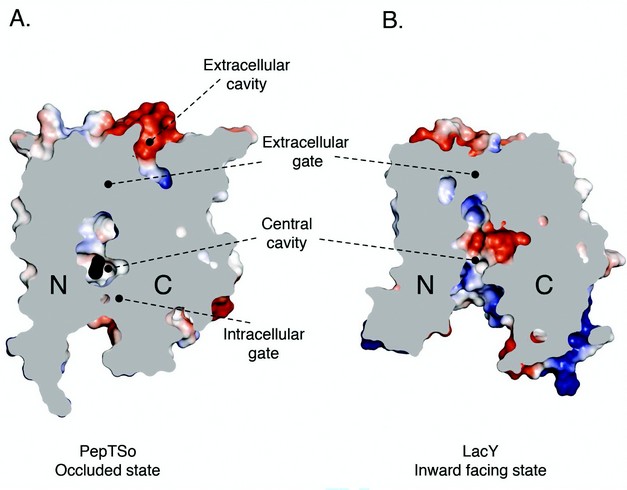
Figure 2: Comparison of occluded PepTSo and open LacY structures. Electrostatic surface representation showing the location of the hydrophilic cavities in a section through the protein volumes of (A). PepTSo and (B). LacY. The N- and C- terminal six helix bundles are labeled. Bound ligand is shown as a black CPK model of a di-alanine peptide. Helices HA and HB have been omitted for clarity.
Although the primary structure, tissue distribution and substrate specificity have all been investigated for mammalian peptide transporters, there is currently no structural information on these proteins, which has severely hampered further investigation into the molecular mechanism of peptide transport and the utilisation of these proteins as ‘Trojan horses’ for drug delivery. To gain much needed insight into the molecular mechanism of transport we determined the crystal structure of a prokaryotic member of the POT family, from the bacterium Shewanella oneidensis, PepTSo (Fig. 1).5 PepTSo shows a high degree of sequence conservation within the transmembrane region (~30 % identity) to the mammalian PepT1 and PepT2 proteins and associated functional data reveal similar kinetic properties supporting a common mechanism of peptide transport. PepTSo contains 14 transmembrane helices, of which helices H1-H12 adopt the overall fold observed previously in other members of the MFS transporter superfamily, with the N- and C-terminal six helical bundles, formed by helices H1-H6 and H7-H12, coming together to form a V-shaped transporter, with both halves related by a pseudo-two fold symmetry axis running normal to the membrane plane. PepTSo is the largest member of the MFS family for which a crystal structure is available, with the additional two helices being formed from the large intracellular loop connecting the end of helix H6 to the start of helix H7. This raises interesting questions as to the role of these additional helices in the biochemistry of bacterial peptide transporters and more generally within the MFS.
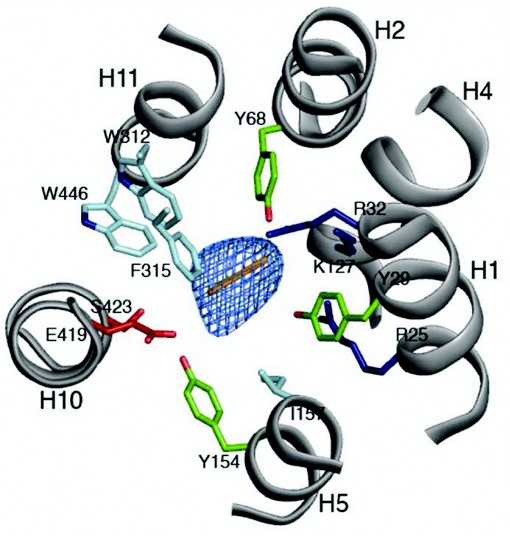
Figure 3: The peptide binding site. The central cavity in PepTSo as viewed from above on the extracellular side of the membrane. Conserved residues between PepTSo and the mammalian peptide transporters are labeled and coloured according to side chain type, Arg and Lys (blue), Glu and Ser (red), Tyr (green) and Trp, Phe and Leu (cyan). A di-peptide sized Ca baton (orange) is fitted as a size reference only into the mFo-DFc electron density observed in the central cavity (blue mesh), contoured at 4 s.
The structure revealed a previously unobserved ligand bound occluded state, with an unidentified ligand bound within a large central cavity that is closed to both side of the membrane (Fig. 2). Many of the residues conserved between PepTSo and the mammalian peptide transporters cluster around this cavity, which has approximate dimensions of 13 x 12 x 11 Å. The cavity is striking for the number and placement of charged residues that cluster to opposite ends to create a potential macro dipole across the two halves of the transporter (Fig. 3). This orientation of charged residues within the binding site suggests a possible mechanism of peptide recognition, with the amino terminus of peptides interacting with the conserved negative glutamic acid (E419) on the C-terminal six helical bundle and the carboxy termi interacting with the conserved cluster of positively charged arginines and lysine residues (R25, R32 and K127) on the opposing, N-terminal bundle. The additional placement of three conserved tyrosine residues is also reminiscent of many antigen recognition sites, where tyrosines play pivotal roles in providing a flexible, hydrophobic environment for side chain accommodation. These characteristics could also explain the specificity we observe within the POT family for di- and tri-peptides, as the dimensions of the cavity are only sufficient to accommodate peptides of these lengths, with larger tetra-peptides being sterically restricted and single amino acids unable to interact with both halves of the transporter and so presumably fail to trigger the required conformation changes to re-orientate the binding site across the membrane.
The occluded conformation reveals many intriguing new features concerning possible mechanisms for transport, both generally within the MFS superfamily and more specifically within the POT peptide transporters. The most obvious of these is the potential significance of the extracellular cavity we observe opening out towards the extracellular side of the membrane. The cavity causes PepTSo to adopt an overall asymmetric state, breaking the pseudo two-fold symmetry observed in previous MFS structures. We are currently working to determine the significance of this with respect to specific conformational changes within the C-terminal helical bundle during transport. The structure however does suggest that within the POT family at least, it is the C-terminal six helical bundle that may contain the mobile gates, both extracellular and intracellular, that alternately open and close around peptides to move them across the membrane. Interestingly, observed at the base of the extracellular cavity is a conserved histidine residue, shown to be protonated during the transport cycle in PepT1. Mutating this residue in PepTSo also abolishes transport; raising the question as to what role both this residue and the additional cavity play in the mechanism of peptide transport.
In summary, there is a pressing requirement to understand the molecular mechanism of human peptide transporters. Their central role in human health, dietary absorption and their effect on drug disposition make them promising targets to improve the pharmacokinetic properties of many drug molecules. The structure of PepTSo provides the first structural model with which to further investigate the underlying biochemistry of these processes and gain further insight into the transport of peptides and drugs across cellular membranes.
Newstead, S., Drew, D., Cameron, A.D., Postis, V.L.G., Xia, X., Fowler, P.W., Ingram, J.C., Carpenter, E.P., Sansom, M.S.P., McPherson, M.J., Baldwin, S.A., Iwata, S. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J 30, 417-426 (2011)
References
Funding Acknowledgments
The BBSRC, Membrane Protein Structure Initiative (MPSi) (grant BBS/B/14418), with important contributions from the Wellcome Trust (grant 062164/Z/00/Z) and the Japan Science and Technology Agency. We wish to thank the Membrane Protein Laboratory (MPL) staff at the Diamond Light Source Limited for their support. We also wish to thank the beamline staff at Diamond Light Source beamlines I02, I03 and I04 and at the European Synchrotron Radiation Facility, beamlines ID23, ID29 and ID14eh4.

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.