The structural basis for catalysis and substrate specificity of a rhomboid protease
Jun 2, 2011
Jun 2, 2011
Rhomboids belong to the family of intramembrane proteases that use a catalytic dyad of serine and histidine for proteolysis of substrate transmembrane proteins. Conserved in both prokaryotes and eukaryotes, they regulate diverse cellular processes such as intercellular signalling, parasitic invasion of host cells, and mitochondrial morphology. Their importance in biology provides a strong incentive to understand the mechanism of these unusual enzymes for identification of specific inhibitors. We obtained the structure of the E. coli rhomboid GlpG covalently bound to a mechanism based isocoumarin inhibitor. The position of the oxyanion hole, as well as the S1 and S2’ substrate binding subsites of GlpG can be inferred from the structure. Subtle structural change is observed upon inhibitor binding and is probably required for catalysis. The structure with the bound inhibitor provides a template to model substrate binding at the active site and thereby lay foundation for a structural explanation of rhomboid specificity and mechanism, as well as for inhibitor design.
Recent identification of intramembrane proteases has revealed a new strategy for cellular regulation where transmembrane (TM) proteins inactive in their membrane bound form are activated by intramembrane proteolytic cleavage. Proteolysis results in the release of cytoplasmic, luminal or extracellular domains that move to new locations where they can carry out their biological function. Different intramembrane protease families, classified as serine, aspartyl or metalloprotease have been identified in all kingdoms of life. Intramembrane proteolysis has so far been implicated in diverse cellular processes including differentiation, the unfolded protein response, lipid metabolism, signal peptide processing and, in prokaryotes, generation of peptide pheromones, response to extracellular stress and pathogenesis.1,2
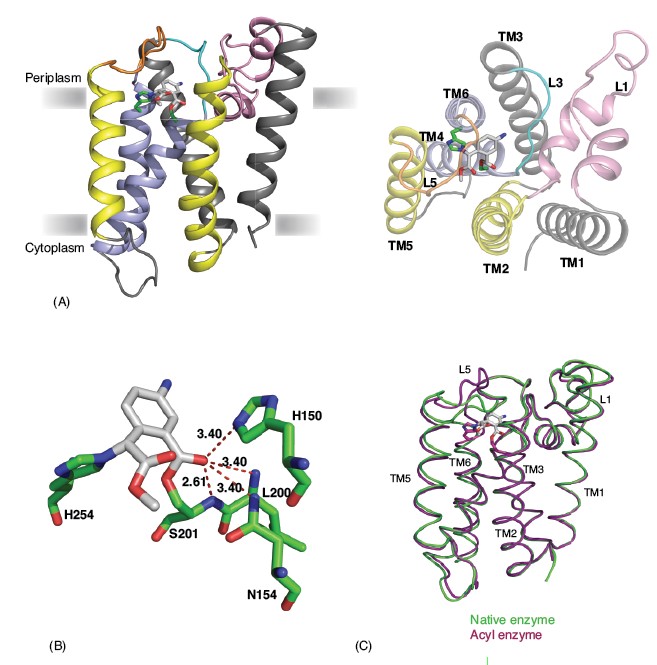
Figure 1:
Rhomboids are six TM domain serine proteases that were identified originally as a component of the EGF receptor-signalling pathway in Drosophila and named after the appearance of mutant embryos. In vivo and in vitro cleavage assays of different rhomboids have identified conserved serine-histidine dyad as key residues for catalysis. Since the hydrolysis of peptide bonds requires water, the concept of proteolysis in the membrane has remained uncertain, and in particular the question of whether the active site resides within the membrane or at the surface where water molecules are plentiful. The crystal structure of prokaryotic rhomboids from Escherichia coli and Haemophilus influenzae reveal the global architecture and show how the catalytic dyad is sequestered in the bilayer.3,4,5,6 In the core, a relatively short TM helix 4 and an extended loop 3 is surrounded by other helices to create a hydrophilic cavity that opens to the periplasmic side. The active site serine is located ~ 10 Å into the bilayer and hydrogen bonded to the conserved histidine. Numerous water molecules are found in this cavity that is capped by flexible loop 5. It is likely that this loop can flip spontaneously. Despite the structural and biochemical advances, the lack of any structure of enzyme-substrate or enzyme-inhibitor complexes leaves important questions to be answered.
As an initial step in understanding the mechanism of rhomboid protease, we determined the structure of E.coli rhomboid GlpG covalently bonded to an isocoumarin inhibitor through both the active site serine and histidine. The inhibitor lies on the active site, ~4.5Å below the plane of the lipid head groups in the membrane (Fig. 1A). The amino group of the inhibitor points towards loop 3, while the methoxy group of the inhibitor points towards the gap between TM2 and TM5 and the lipid bilayer. The catalytic efficiency, as defined by increased rate constant or decreased transition state energy, of classical serine proteases is enhanced by the so-called oxyanion hole. The benzoyl carbonyl oxygen of amino-methoxy-isocoumarin mimics the substrate peptide carbonyl, allowing us now to identify its position as the oxyanion hole in GlpG, and to identify the probable residues involved. There are four potential hydrogen-bonding donors in the vicinity of the benzoyl carbonyl oxygen. These include the main chain amides of Ser201 and Leu200, the side chain amide of Asn154 and the nitrogen (Ne) from the imidazole ring of His150, highly conserved residues in rhomboids. In the present structure the main chain amide of Ser201 forms a strong hydrogen bond with the benzoyl carbonyl oxygen of the inhibitor, while the other residues form hydrogen bonds that can be inferred from their length to be weaker (Fig. 1B). With the structure of an alkylated acyl enzyme of GlpG, we now demonstrate unequivocally that rhomboids are serine proteases that use a catalytic mechanism analogous, but not identical, to the classical soluble serine proteases.
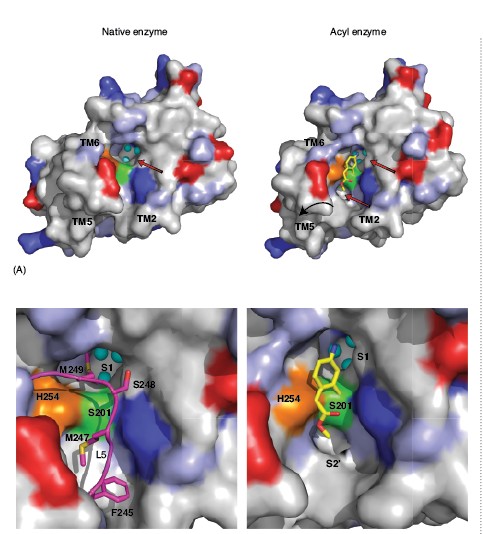
A comparison of the inhibitor-bound acyl enzyme structure with that of the native enzyme without a substrate or inhibitor bound reveals several significant structural changes (Fig. 1C). In the native enzyme structure, loop 5, with two bulky methionine side chains (Met247 and Met249), caps the active site. In the acyl enzyme structure we observe that loop 5 is lifted away from the active site, consistent with the view that it occludes the catalytic centre in the resting state of the enzyme. Accompanied with this movement of loop 5 is a slight displacement of TM5 and TM6, which flank loop 5. A requirement of structural change in TM5 can be appreciated from the surface analysis of the rhomboid acyl enzyme structure, which reveals two cavities on either side of the bound inhibitor (Fig. 2). The first of these cavities is in close proximity to the active site and the amino group of the inhibitor points into it. This cavity is also observed in the native structure of rhomboid, and is filled with three water molecules. In the native enzyme structure the side chain of Met249 from loop 5 plugs this cavity (Figs. 2A and B). The second cavity observed only in the acyl enzyme structure is located on the opposite side of the catalytic dyad and the methyl acetate group of the inhibitor points towards it. It is not fully formed in the native enzyme structure, where it resembles a shallow cleft (Fig. 2A). Thus the inhibitor bound structure suggests that a small movement of TM5 along with loop 5 is essential for substrate binding and catalysis. Interestingly, this conclusion, challenges earlier models that included major TM5 movement during catalysis.
Grooves and cavities in an enzyme structure often represent substrate binding subsites, where the side chains of amino acids from a substrate can interact and thereby the specificity for a given substrate can be achieved. Based on the biological and biochemical considerations, we propose that the cavity in GlpG where the amino group of the inhibitor points is the S1 subsite of GlpG and the second cavity, which is observed only upon inhibitor binding and its relative distance from the catalytic residues leads us to propose that this could represent the S2’ subsite (Fig. 2). The identification of substrate binding sites in GlpG structure not only allows modelling a short peptide at the active site but also for inhibitor design. The architecture of the active site of GlpG covalently bound to an isocoumarin provides an important foundation for the design and optimisation of inhibitors for therapeutic opportunities but also as a research tool to uncover the intermediate steps in the mechanism of rhomboids.
Vinothkumar, K.R., Strisovsky, K., Andreeva, A., Christova, Y., Verhelst, S., Freeman, M.The structural basis for catalysis and substrate specificity of a rhomboid protease. EmboJ. 29, 3797-3809
References
Acknowledgements
Medical Research Council, UK and Marie-Curie Intraeuropean Fellowship.
Research carried out at Diamond Light Source on IO2/IO3/IO4 and SLS X06SA.

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.