 | ||
Visualising how nature uses vitamin B12 to deal with halogenated pollutants |
Beamline I04 Scientific Highlight
Structure determination using the Macromolecular Crystallography (MX) beamlines at Diamond, and beamline I04 in particular, combined with electron paramagnetic resonance spectroscopy and computational simulations showed a direct interaction between the cobalamin cobalt ion and the halogen substrate. Previously studied vitamin B12-dependent enzymes catalyse carbon-cobalt bond formation, however, it is proposed that the reductive dehalogenases act as catalysts via halogen-cobalt bond formation. A halogencontaining material binds to the cobalt in cobalamin, followed by a reduction that results in halogen removal from the substrate. These results present a new model in organohalide and cobalamin chemistry that will guide future research into the exploitation of this group of enzymes, in biocatalysis or in the removal of halogenated pollutants from the environment.
The last two decades have seen a great increase in our understanding of enzymes that use vitamin B121,2. As one – if not the – most complex of cofactors, vitamin B12 is an intricate organometallic molecule, consisting of a cobalt (Co) ion coordinated by a tetrapyrrole-derived macrocycle (the corrin ring)2. This cofactor has many unique properties: the Co(I) species is highly nucleophilic, while the Co-C bond in the Co(III)-carbon ligated species can be homolytically (giving rise to a Co(II) species and a carbon radical) or heterolytically cleaved depending on the enzyme. Hence, the B12-dependent enzymes can be classified according to the type of reaction catalysed: Group I consists of the 5’-deoxyadenosyl B12 (AdoCbl) dependent enzymes that catalyse intramolecular 1,2-rearrangements using AdoCbl as a reusable radical via homolytic cleavage of the Co-C bond. Group II contains the corrinoiddependent methyltransferases that use methylcobalamin, which in contrast to the AdoCbl using enzymes undergoes heterolytic cleavage to methylate the enzyme substrate with a methyl cation, cycling the Cbl cofactor between cob(I) alamin and methylcob(III)alamin redox states. Group III contains the reductive dehalogenases, which use a corrinoid cofactor and two iron-sulfur groups to catalyse a reductive dehalogenation3.
In contrast to group I and II, little is known about the structure or mechanism of the group III reductive dehalogenases. This is due to the fact that these enzymes have proven difficult to isolate in larger quantities, while numerous attempts to heterologously produce these enzymes have so far failed. The ability to dehalogenate both aliphatic and aromatic substrates at specific positions is, however, of considerable interest, not only from a mechanistic viewpoint, but also in view of the role these enzymes play in the global halogen cycle4. This cycle is now heavily impacted upon by industrial halogenated waste and these enzymes have obvious application in bioremediation or biocatalysis as effective dehalogenases. Following a concerted effort over a five-year period, the research group has managed to heterologously produce an active reductive dehalogenase in Bacillus megaterium5.
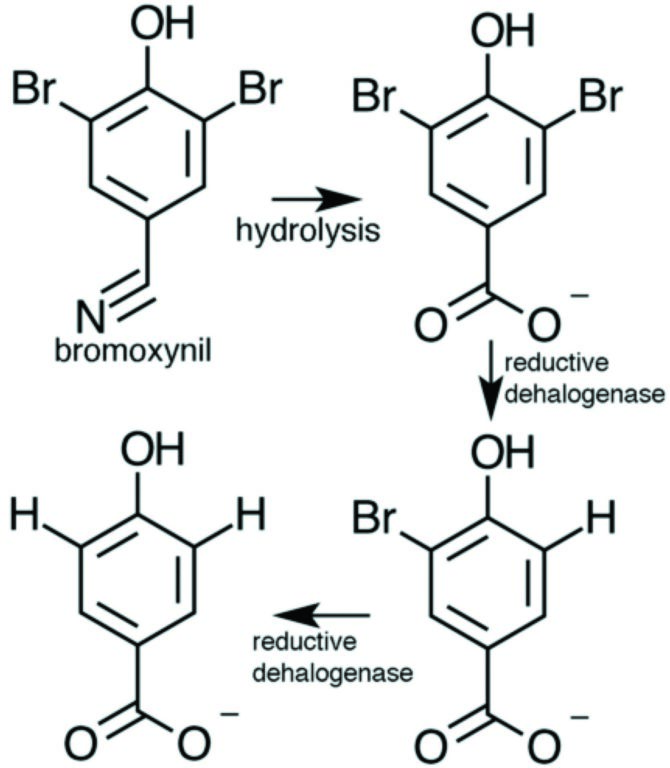
Figure 1: Reaction catalysed by the reductive dehalogenase from Nitratireductor pacificus. Following hydrolysis of the herbicide bromoxynil by other enzymes, the dehalogenase removes the bromide atoms in a step wise manner.
While the majority of reductive dehalogenases are membrane-associated and oxygen-sensitive enzymes that are key to organohalide respiring bacteria3, a sub-class of soluble and oxygen-tolerant enzymes has recently been found. The latter serve in a catabolic rather than a respiratory role and are found in non-organohalide respiring bacteria5. The group have cloned and expressed several representatives of both reductase classes in E.coli and B. megaterium, and found a gene encoding for a putative dehalogenase from Nitratireductor pacificus (RdhNP) to yield mg/L quantities of active protein in the Bacillus host. Kinetic studies indicate the enzyme can reductively debrominate and dechlorinate ortho-halogenated phenol groups, with the preferred substrate being 3,5-dibromo-4-hydroxy-benzoic acid (Fig. 1). This component is a known breakdown product of the widely used nitrile herbicide bromoxynil. Using data collected at IO4, but also at other Diamond beamlines, the group solved the crystal structure of the 70kDa RdhNP using the inherent, but weak Fe anomalous signal. The 2.3 Å structure reveals the corrinoid is bound in the base-off conformation, in close proximity to one of the two 4Fe4S clusters (Fig. 2). This particular state is uncommon, and has previously been associated with raising the midpoint potential of the Co(I)/Co(II) pair from -660mV to ~-350mV, making it accessible to physiological reductants2.
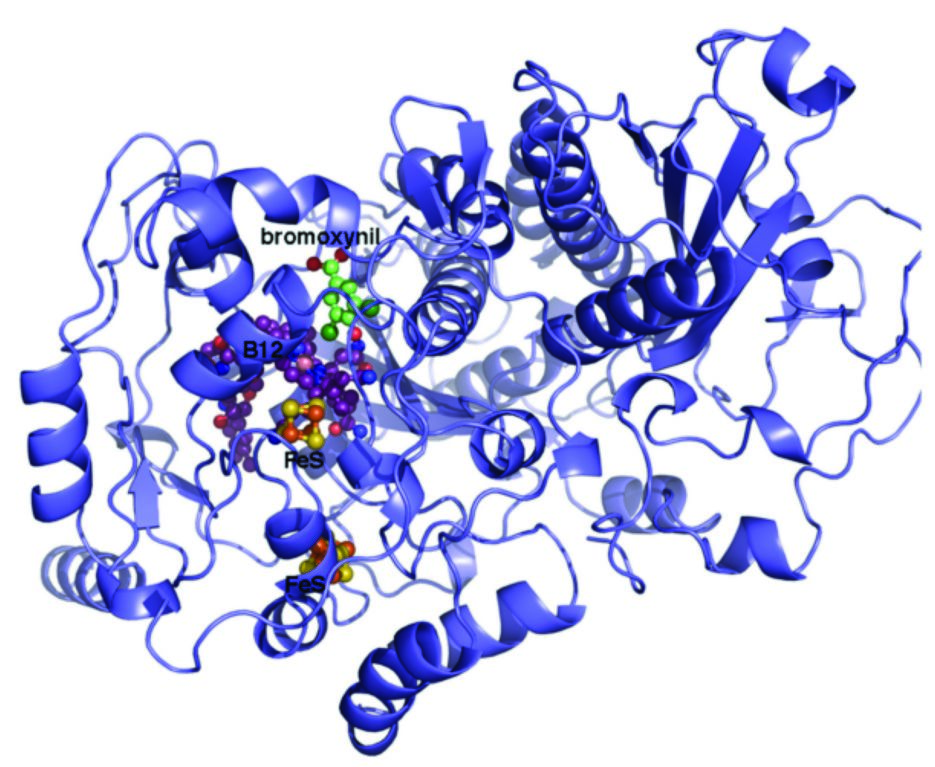
Figure 2: Crystal structure of the reductive dehalogenase determined to 2.3 Å. The bound FeS and B12 cofactors and bromoxynil derived substrate are shown in atom coloured spheres while the protein is shown in cartoon representation.
Analysis of the active site architecture, and the conservation of residues lining the active site suggests an unprecedented mechanism for reductive dehalogenases. While abiotic reductive dehalogenation by B12-like molecules can be observed (using low potential reductants such as titanium citrate), the mechanism of this reaction has been alternatively proposed to be either radical based or through transient formation of a Co-C substrate bond. The present literature on reductive dehalogenases has proposed similar mechanisms might occur for the biotic reaction3. However, the group's analysis of RdhNP active site structure apparently rules out the possibility of a transient Co-C complex. Furthermore, the solvent exposed nature of the active site, combined with the oxygen-tolerant nature of this particular enzyme, is in the group's opinion incompatible with a radical reaction. In contrast, the ligand-enzyme complex model suggests a mechanism whereby the highly nucleophilic Co(I) species attacks the halogen atom directly (for example, an oxidative addition), concomitant with a proton donated from the conserved Tyr426 to the substrate carbon (Fig. 3). In many ways, the proposed reductive mechanism is similar to the well-established oxidative attack carried out by the heme containing P450 enzymes. In the latter, the highly oxidative compound I is made following substrate and oxygen binding through two separate electron transfer events, and substrate is hydroxylated at position(s) closest to the compound I species. In the reductive dehalogenases, we propose substrate binding gates the formation of the highly nucleophilic Co(I) species, which through oxidative addition reduces the substrate at positions closest to the corrinoid Co.
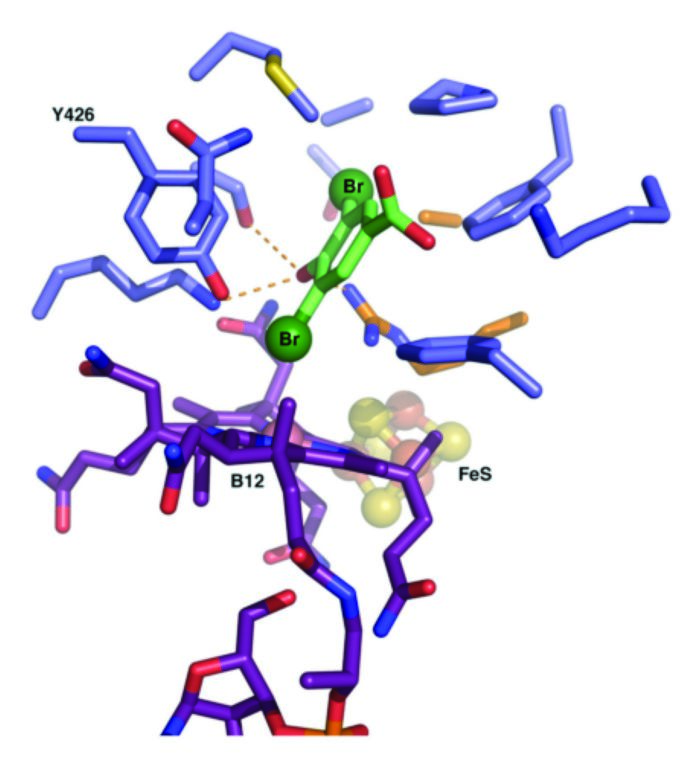
Figure 3: Modelled complex of the enzyme with the halogenated substrate. One of the bromide atoms is proposed to contact the B12 cobalt ion.
Source publication:
Payne, K. A. P., Quezada, C. P., Fisher, K., Dunstan, M. S., Collins, F. A., Sjuts, H., Levy, C., Hay, S., Rigby, S. E. J. & Leys, D. Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature 517, 513-516, doi:10.1038/nature13901 (2015).
References:
1. Banerjee, R. & Ragsdale, S. W. The many faces of vitamin B-12: Catalysis by cobalamin-dependent enzymes. Annual Review of Biochemistry 72, 209- 247, doi:10.1146/annurev.biochem.72.121801.161828 (2003).
2. Brown, K. L. Chemistry and enzymology of vitamin B-12. Chemical Reviews 105, 2075-2149, doi:10.1021/cr030720z (2005).
3. Smidt, H. & de Vos, W. M. Anaerobic microbial dehalogenation. Annual Review of Microbiology 58, 43-73, doi:10.1146/annurev. micro.58.030603.123600 (2004).
4. Oberg, G. The natural chlorine cycle - fitting the scattered pieces. Applied Microbiology and Biotechnology 58, 565-581, doi:10.1007/s00253-001- 0895-2 (2002).
5. Payne, K. A. P. et al. Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature 517, 513-516, doi:10.1038/nature13901 (2015).
Funding acknowledgement:
This research was funding by the European Research Council, DEHALORES206080.
Corresponding author:
Professor David Leys, University of Manchester, [email protected]
