Targeted Destruction of Disease-Related Proteins
Mar 17, 2025
Mar 17, 2025
While most conventional drugs work by inhibiting proteins, not all proteins are easy to block in this fashion. Drug developers are investigating new classes of drugs that mark proteins for degradation in the cell. A large, barrel-shaped structure called the proteasome drives this breakdown process, and a protein called Cereblon behaves as an usher, delivering proteins to the proteasome for destruction. Some drugs act as “molecular glue”, sticking to Cereblon and altering its structure so that it binds to target proteins. Other drugs called proteolysis targeting chimeras (PROTACs) bind to target proteins and Cereblon, bridging the two together. Thus, an in-depth understanding of Cereblon's morphology is crucial for drug investigations. However, scientists have struggled to determine high-resolution structures of this protein in the past due to complications with its synthesis and stability. David Zollman, a structural biologist and drug developer at the University of Dundee, and his colleagues developed a highly stable, easily purified Cereblon variant. Collecting X-ray crystallography data at the Diamond Light Source beamlines I04 and I24, they demonstrated that the structure of their Cereblon variant matched ones previously collected by other groups, but the new crystals achieved higher resolution. Cereblon changes shape when bound to different drugs, and the team collected small-angle X-ray scattering (SAXS) data at beamline B21 to study how shapeshifting varies between different drug candidates. Together, these findings reveal that the new Cereblon variant is amenable to structural analysis, which could facilitate future research into this promising class of protein-degrading drugs.
Most conventional drugs work by inhibiting proteins. The pain-reliever ibuprofen, for example, blocks a bodily enzyme called cyclooxygenase by stoppering its active site and preventing it from producing chemical signals that induce pain. However, Zollman said that researchers have long considered some proteins “undruggable” because they lack active sites that can be targeted by inhibitors. These include proteins that have structural roles rather than enzymatic functions. Taking an alternative approach, scientists are exploring drugs that flag proteins for degradation in the cell by protein shredders called proteasomes.
The most infamous example is the drug thalidomide, a sedative from the 1950s that pregnant women took to relieve morning sickness but led to birth defects. Today, doctors have repurposed thalidomide to treat multiple myeloma, and researchers have developed other drug candidates, like lenalidomide and mezigdomide to treat other cancers. Currently, there are over 40 drugs related to the degradation pathway in cells undergoing clinical trials. Many of them work by recruiting transcription factors to Cereblon and targeting them for destruction, thereby preventing the expression of an array of genes.
Research into these drugs has been held back by a lack of structural insight into Cereblon. Previously, scientists could only purify Cereblon coupled with an adapter protein called Damage Specific DNA Binding Protein 1 (DDB1), resulting in an unwieldy complex. Scientists also struggled to produce high yields of the protein, and they could only prepare it in insect cell expression systems. When scientists managed to crystallize the protein, they found it was unstable, hampering efforts to collect high-resolution structural data. Most experiments determined the structure to a resolution of 3 Ångströms (Å) or worse. Dr Zollman said:
It’s expensive to produce, hard to get in large quantities, and then when you do have it, it’s quite poorly behaved.
What scientists needed was a stable version of Cereblon that was easy to purify in the absence of DDB1. Dr Zollman commented:
We have cut out the part of Cereblon that binds to DDB1, and because of that, we are able to produce it stably from E. coli on its own.
E. coli are the go-to bacteria for producing proteins for purification, making it easier to achieve high yields for scientific studies.
Besides omitting the DDB1-binding domain, Zollman's team designed 15 versions of Cereblon, some of which carried unique sets of mutations that swap out one amino acid for another in different places. They introduced these mutations to stabilize the proteins, and they discovered that version 8, complete with 12 mutations, proved most stable. “We can get it at a much higher yield, it’s much cheaper to produce, it’s much easier to produce, and then the complex does crystallize a lot better.” Zollman said version 8 is a “middle ground” between full-length Cereblon and other truncated versions trialled previously, so his team renamed it Cereblonmidi.
Next, they had to put their crystallised Cereblonmidi to the test at the Diamond Light Source. Zollman said the protein formed small crystals, and the microfocus beams at beamlines I04 and I24 enabled his team to collect high-quality data from samples of this size.
They investigated Cereblonmidi bound to mezigdomide and lenalidomide to a higher resolution than was previously possible: 2.19 and 2.50 Å, respectively. They were able to observe some never-before-seen features, such as interactions between a fluorine atom on the drugs with an aspartate residue on the protein. Dr Zollman said:
If you overlay the structure with equivalent cryo-electron microscopy or crystal structures that have been published before, we do have extremely good alignment. Where there are differences, it tends to be because those regions are poorly resolved in the original structures, and we are actually able to see them in a clearer way.
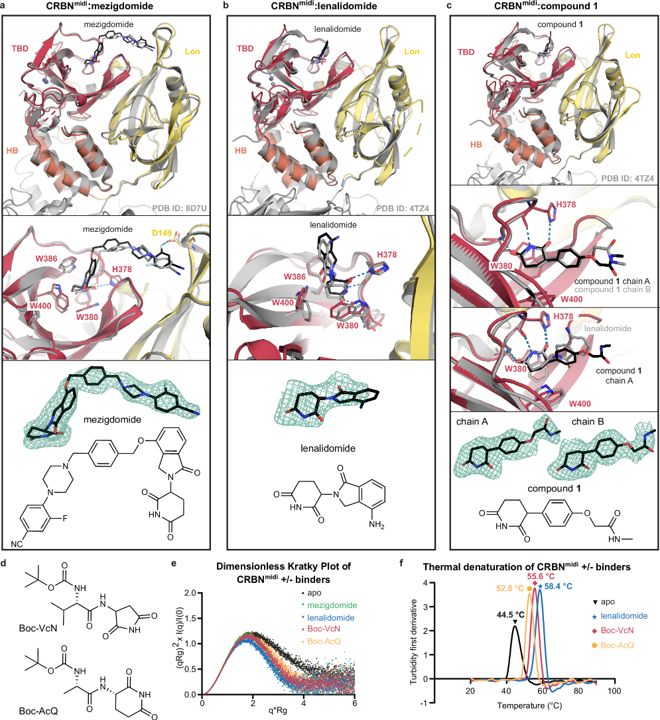
Cereblon exists in an open conformation, like an open book, when it doesn’t bind to a drug and a closed conformation, like a shut book, when it latches onto one. Cereblonmidi only crystallized in the closed conformation, so the team needed to study the protein in solution to track its opening and closing. SAXS experiments at the B21 beamline allow scientists to determine the overall 3D structure of protein complexes in solution, and the team tested out how multiple drugs affect the shape of Cereblonmidi using this technique. When they collected SAXS data from unbound protein, they found that the protein swung open, but when lenalidomide or mezigdomide bound it swung closed. This suggested that SAXS could confirm if other drug candidates bind to Cereblonmidi and affect its shape. They team tested out small molecules called Boc-protected cyclic-immunomodulatory di-peptides that mimic Cereblon’s natural substrates, finding that they also closed the protein. “Cereblon is a scavenger of damaged proteins in the cell,” Zollman noted. “Its native function is to look for proteins that have been cleaved at unexpected residues,” and these di-peptides mimic those damaged proteins.
Nathan Cowieson, Principal Beamline Scientist at B21, added:
Drugs that interrupt protein-protein interfaces or cause large conformational changes can be really challenging to study by crystallography as they will tend to destroy crystals. This study is a great demonstration of what a useful tool SAXS can be alongside crystallography in these challenging cases.
Lastly, the team returned to X-ray crystallography to see if they could capture Cereblonmidi in complex with a drug as well as its target protein. They succeeded at capturing Cereblonmidi stuck bound to mezigdomide and a target transcription factor called zinc-finger of neo-substrate Ikaros (IKZF1ZF2), which has been implicated in cancer and autoimmune disorders. This will provide novel insight into the mode of action of this experimental treatment.
Zollman said there are hundreds of research groups working on developing drugs that target proteins for degradation. “A lot of these drug discovery programs may well have been hindered by not having high-resolution insight.” Now, his team have developed a new variant suitable for structural work, which they hope others will leverage to bolster drug research in the future.
Some drugs stick to proteins and target them for degradation by the cell. The protein Cereblon is pivotal to this feat, but researchers have struggled to capture its structure in detail. Users at beamlines B21, I04, and I24 used small-angle X-ray scattering and X-ray crystallography to show that a new Cereblon mutant could be captured at a higher resolution, bolstering efforts to study how new drugs interact with this protein.
To find out more about B21 or discuss potential applications, please contact principal beamline scientist Nathan Cowieson: [email protected].
To find out more about I04 or discuss potential applications, please contact principal beamline scientist Ralf Flaig: [email protected].
To find out more about I24 or discuss potential applications, please contact principal beamline scientist Robin Owen: [email protected]
Kroupova A, et al. Design of a Cereblon construct for crystallographic and biophysical studies of protein degraders. Nature Communications. 15, 8885 (2024). DOI:10.1038/s41467-024-52871-9
Images credits: figure: this publication 10.1038/s41467-024-52871-9 under CC BY 4.0 license.
for the thumbnail: The image is sourced from Dundee university

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.