The Hedgehog signalling pathway is one of the many ways cells communicate with each other, and it is involved in embryonic development, cancer genesis and stem cell function. The structure of a protein which helps transmit these signals across a cell membrane has been solved using the tuneable Microfocus beamline (I24) and the High Throughput Small Angle X-ray Scattering (SAXS) (B21) beamline at Diamond Light Source. The protein, Smoothened, is a G-protein-coupled receptor (GPCR), spanning the cell membrane and protruding beyond it. Researchers used I24 to generate diffraction data from small crystals (no larger than 10-20 μm long) of the protein, and B21 to understand the dynamics and large-scale domain reorientations of the protein in solution.
Diffraction data collected at I24 revealed the crystal structure of Smoothened, including its large extracellular domain. This is the first time the protein structure of a GPCR with a large extracellular domain has been decoded. The researchers also found a cholesterol molecule bound to Smoothened’s extracellular domain, a discovery that might be the long sought-after endogenous small molecule that regulates Hedgehog signalling. The crystal structure of Smoothened bound to a common anti-cancer drug was also determined. Mapping the structure of Smoothened will aid researchers in understanding why certain mutations impact drug binding and support efforts to combat resistance to anti-cancer drugs.
Smoothened (SMO) is a transmembrane protein that serves to transmit signals of the Hedgehog (Hh) pathway, a cell-cell communication system crucial for the development of most tissues in vertebrates, including humans. SMO activity is unleashed when its upstream negative regulator, Patched, is inactivated by extracellular Hh ligands, allowing SMO to convey the Hh signal across the membrane to the inside of the cell. This signal transduction step is most commonly damaged in Hh-driven human cancers but the mechanistic details behind it remain unknown.
Several anti-cancer drugs directly inhibit SMO in order to switch off the Hh pathway and consequently stop the growth of tumours, such as basal cell carcinoma and medulloblastoma, that are addicted to unrestrained Hh signalling in the longest dimension. However, these cancers can develop resistance by mutating the site within SMO at which the drugs bind. A detailed picture of how SMO works normally and how these drugs bind to SMO may hold the key to developing better anti-cancer drugs.
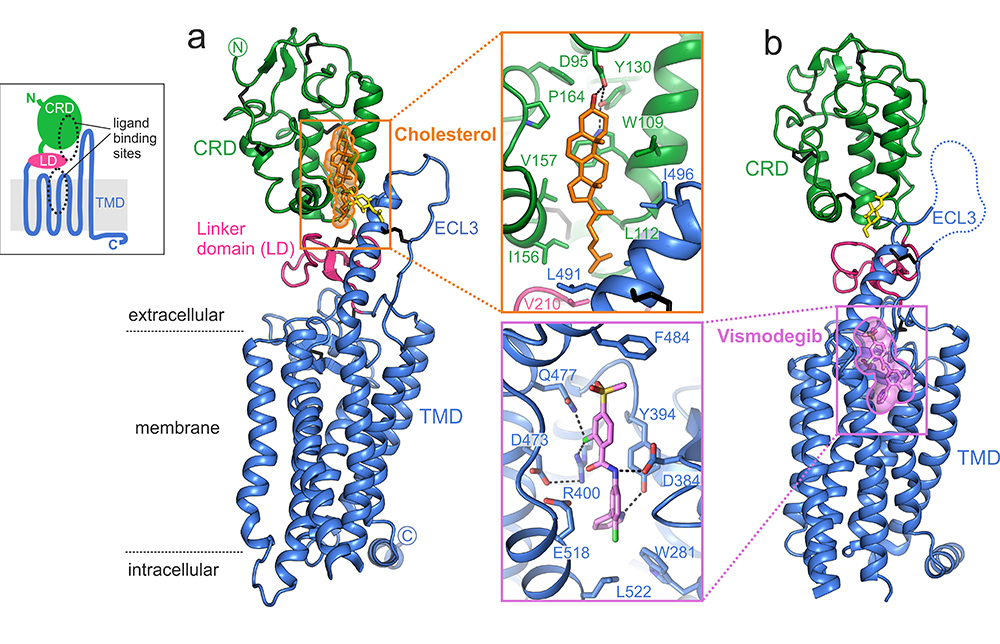
Figure 1: Structure of human SMO. (a), Overall structure of human SMO showing full extracellular and transmembrane domains (cartoon) in complex with cholesterol. (b), Overall structure of human SMO (cartoon) in complex with anti-cancer drug vismodegib. Insets show close-ups of cholesterol (top) and vismodegib (bottom) with interacting residues as sticks. Green: CRD; pink: linker domain; blue: TMD; orange: cholesterol; violet: vismodegib; yellow sticks: N-linked glycans (NAG); black sticks: disulphide bonds; black dotted lines: potential hydrogen bonds.
While most GPCRs are activated or inhibited via a single small-molecule binding site located in their transmembrane domain (TMD), SMO has two small-molecule binding sites: one in the TMD and another in its large extracellular cysteine-rich domain (CRD), both essential for proper signalling function1. However, in the absence of a crystal structure containing both domains, the positioning and interactions between these two sites have remained unclear.
To address these questions, the structure of SMO was solved including both its CRD and TMD (Fig. 1). A transient mammalian expression system, combined with antibody affinity purification and lipidic cubic phase (LCP) crystallisation, was used to produce microcrystals of SMO. These microcrystals were too small for conventional beamlines (~10-20 μm) and thus necessitated the use of the B21Microfocus Macromolecular Crystallography beamline (I24) at Diamond Light Source in order to obtain diffraction data. The automated ‘grid-scan’ software function at I24 was also essential for screening a great number of visibly opaque LCP drops to find well-diffracting crystals. The merging of data from multiple crystals was required to obtain a full dataset and ultimately solve the structure of SMO to 3.2 Å resolution.
In the structure, the CRD is stacked atop the TMD and separated by the small wedge-like linker domain (Fig. 1a). The two small-molecule binding sites are distinct from one another but appear to interact via the intervening linker domain and the long extracellular extension of TMD helix 6 (extracellular loop 3, ECL3, Fig. 1a). In functional assays, mutating SMO to remove the entire CRD or to disrupt the interaction between the CRD and TMD led to constitutive activation of SMO, suggesting that under normal conditions the presence of the CRD restrains the activity of SMO.
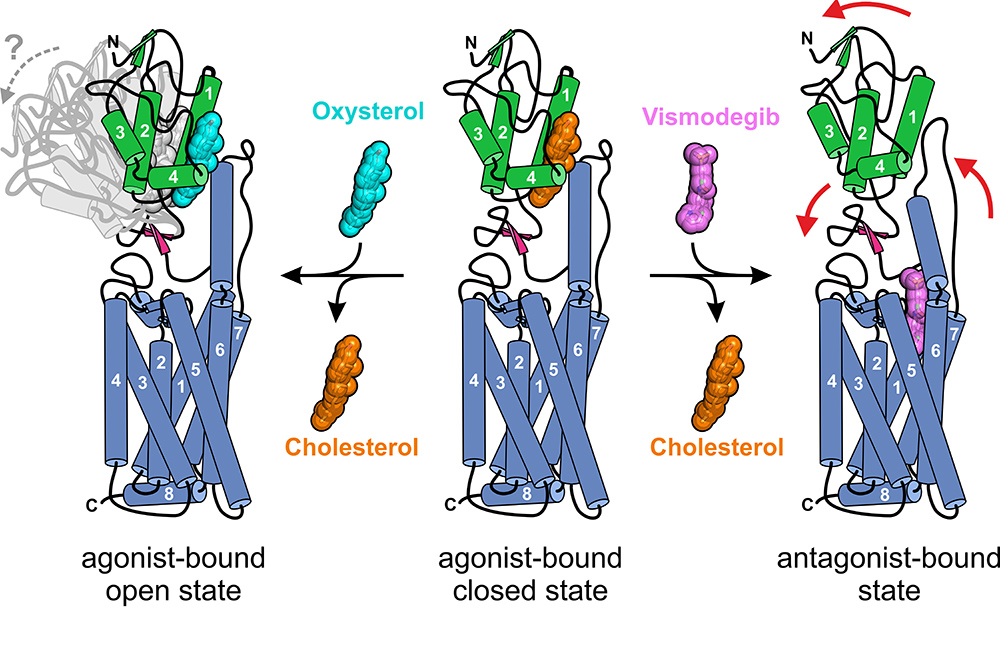
Figure 2: Activation states of SMO with two ligand-binding sites. SMO in complex with cholesterol (middle) represents an agonist-bound but closed state of SMO that can be shifted into the fully-closed antagonist-bound state upon vismodegib binding (right) or into an agonist-bound open state upon oxysterol binding (left), as suggested by SAXS measurements. Colour-coding follows Fig. 1; cylinders represent alpha-helices; red arrows represent conformational changes; cyan: oxysterol
Unexpectedly, a single cholesterol molecule was observed bound to the CRD binding site (Fig. 1a and upper inset). A battery of subsequent experiments revealed that cholesterol can not only bind at the location and in the orientation seen in the crystal structure but also that cholesterol is sufficient, in and of itself, to partially activate the Hh pathway2,3. Cholesterol appears to mediate and stabilise a key interaction between the CRD and TMD. In addition, mutations in the cholesterol-binding site abolished SMO activation in response to native Hh ligands, indicating an essential role for cholesterol in endogenous Hh signalling. This represents the first report of a GPCR that is regulated by the binding of an endogenous small-molecule ligand to an entirely extracellular domain.
To obtain snapshots of SMO in different signalling states, the crystal structure of SMO bound to the antagonist and anti-cancer drug vismodegib was also determined (Fig. 1b). This drug is currently used for the treatment of advanced basal cell carcinoma. Diffraction data were again collected at I24. Vismodegib was bound to the TMD binding site and appeared to induce conformational rearrangements of both ECL3 and the CRD that resulted in loss of cholesterol from the distant CRD binding site (Fig. 2). This work provides the first structural and mechanistic description of an allosteric interaction between two small-molecule binding sites located within two physically-separable domains of a GPCR. This structure also explains the molecular mechanism for various vismodegib-resistance mutations that have been observed in patients’ resurgent tumours4.
Finally, we sought to understand SMO’s activation mechanism through the addition of a small-molecule agonist, a more soluble cholesterol derivative called an oxysterol5, which targets the CRD binding site. Small angle X-ray scattering data of SMO with and without this oxysterol were collected at the High Throughput Small Angle X-ray Scattering (SAXS) beamline (B21) at Diamond Light Source. The inline size-exclusion chromatography plus SAXS capability of B21 was crucial for this experiment because of suspected heterogeneity in the purified protein sample. The data showed that the addition of an activating oxysterol molecule led to a modest overall elongation of the protein molecule, suggesting a conformational movement of the CRD upon activation (Fig. 2, left panel).
Taken together, the two crystal structures provide a molecular explanation for how the CRD and two allosterically linked ligand-binding sites regulate the activity of SMO. The identification of cholesterol as a novel ligand for SMO leads to fascinating questions about the mechanism and evolution of the Hh pathway, while the structure of vismodegib bound to SMO aids the understanding of current anti-Hh drugs and suggests strategies for the design of improved therapeutics. Further investigation will be required to fully understand the conformational changes associated with activation that our SAXS data imply and to determine the nature of Patched’s inhibitory influence on SMO.
References:
- Nachtergaele, S. et al. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife 2, doi: 10.7554/eLife.01340 (2013).
- Luchetti, G. et al. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife 5, doi: 10.7554/eLife.20304 (2016).
- Huang, P. et al. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 166, 1176-1187.e14 doi: 10.1016/j.cell.2016.08.003 (2016).
- Yauch, R. et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 326, 572-574, doi: 10.1126/science.1179386 (2009).
- Nachtergaele, S. et al. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nature Chemical Biology 8, 211-220, doi: 10.1038/nchembio.765 (2012).
Funding acknowledgement:
The work was supported by Cancer Research UK (C20724/A14414), the US National Institutes of Health (GM106078, HL067773), the Wellcome Trust (102890/Z/13/Z, 092970/Z/10/Z and 090532/Z/09/Z), and the Taylor Family Institute for Psychiatric Research. Further support by NDM Oxford, Medical Research Council UK, Ford Foundation and National Science Foundation is acknowledged.
Corresponding author:
Professor Christian Siebold, Wellcome Trust Centre for Human Genetics, University of Oxford, [email protected]
Related publication:
Byrne E, Sircar R, Miller P, Hedger G, Luchetti G, Nachtergaele S, Tully M, Mydock-McGrane L, Covey D, Rambo R, Sansom M, Newstead S, Rohatgi R, Siebold C. Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517-522, doi: 101038/nature18934 (2016).
Publication keywords:
X-ray crystallography; G protein-coupled receptor; Hedgehog signalling; Stem cell regulation; Embryonic development; Smoothened; Oncoprotein; Anti-cancer drug target; Cancer resistance; Cysteine-rich domain; SAXS

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.