Structural basis for retroviral intasome assembly and integrase inhibitor action
Oct 8, 2010
Oct 8, 2010
Dr Peter Cherepanov, Imperial College London
To establish successful infection, a retrovirus must insert DNA replica of its genome into a host cell chromosome. This process is catalysed by integrase (IN), the viral enzyme that synapses ends of viral DNA forming a highly stable nucleoprotein complex, intasome. The structure of full-length IN, either separately or in complex with viral DNA, has been lacking. Furthermore, although clinically useful inhibitors of HIV IN have been developed, their mechanism of action remained speculative. Using data acquired at Diamond beamlines I02 and I04, we determined the long-sought-after crystal structure of a functional retroviral intasome, revealing a tetramer of IN assembled on viral DNA ends. Soaking intasome crystals in the presence of clinical HIV IN inhibitors Raltegravir and Elvitegravir elucidated their common mechanism of action. Binding within the active site, the drugs cause dislocation of the reactive 3’ viral DNA end, disarming the viral nucleoprotein complex. These results represent a quantum leap in understanding of the retroviral DNA integration process and provide a platform for rational design of IN inhibitors.
HIV IN has been aggressively pursued as a target for antiretroviral drug development since early 1990s. Diketo acids, the first class of IN antagonists capable of suppressing HIV spread in cell culture were reported a decade ago [1], and the first clinically-approved HIV IN inhibitor Raltegravir (also known as MK0518 and Isentress) was launched by Merck in 2007 [2]. Drugs with similar modes of action developed by Gilead (Elvitegravir or GS9137) and GlaxoSmithKline (S/GSK1349572) are in advanced clinical trials. Despite its acute importance for antiretroviral drug discovery and decades of rigorous research (reviewed in ref. 3), the complete structure of retroviral IN, either as a separate protein or in the context of the functional nucleoprotein complex (intasome), was lacking. Accordingly, the structural organization of the enzyme active site, predicted to adopt its functional state only upon viral DNA binding, was unknown. Because clinically-useful HIV IN inhibitors bind to and inhibit the intasome as compared to free IN, the mechanism of drug action remained mysterious.
Unfavourable biochemical properties of HIV-1 IN are largely responsible for the paucity of the available structural information. When removed from its natural environment, the enzyme is largely insoluble and poorly active. Crystallography of various IN fragments provided only limited insight into the assembly of its functional complexes. Retroviral IN is comprised of three canonical structural domains. The catalytic core domain (CCD) contains the invariant D,D-35-E motif, which was predicted to coordinate a pair of metal cofactors (Mg2+ or Mn2+). To function, IN further requires its N-terminal domain (NTD), a three-helical bundle stabilised through binding a Zn atom, and a C-terminal domain (CTD) that adopts an SH3-like fold [3]. The domains are connected by flexible linkers, which do not adopt conserved conformations in available partial crystal structures.
Having conducted a wide screen of divergent retroviral INs, we identified the ortholog from the prototype foamy virus (PFV) as an ideal candidate for detailed structural studies [4]. Compared to HIV-1 IN, PFV IN is highly soluble and active in vitro. Furthermore, it is uniquely efficient at synapsing pairs of viral DNA ends and inserting them into exogenous target DNA in a concerted fashion, faithfully reproducing the retroviral integration process in vitro. Importantly, PFV IN is sensitive to clinical HIV IN antagonists and therefore presents a suitable proxy for mechanistic studies of HIV IN inhibitors [4]. Following extensive crystallization trials that involved over 40,000 individual sparse matrix conditions, we identified a crystal form of full-length wild type PFV IN in complex with a 19-bp viral DNA mimic. The crystals diffracted X-rays to 2.9 Å resolution, enabling us to determine the three-dimensional structure of the intasome. The asymmetric unit contained a single IN dimer with a tightly associated viral DNA molecule, and a pair of symmetry-related IN dimers formed the intasome (Fig. 1).
The architecture of the complex is drastically different from previously reported models, which were all based on partial IN crystal structures. In total, almost 10,000 Å2 of molecular surface is buried within IN-DNA interfaces of the intasome. The protein-DNA interactions involve amino acid residues from each domain of the inner IN subunits, their inter-domain linkers, and 17 nucleotides from each viral DNA end. The canonical retroviral IN domains (NTD, CCD and CTD) do not have discrete functions; each contributes to extensive protein-protein and protein-DNA contacts within the functional nucleoprotein complex. The reactive 3′ termini of the DNA molecules are positioned within close proximity of the Asp-128, Asp-185 and Glu-221 active site carboxylates, which collectively comprise the D,D-35-E motif in PFV IN. Soaking intasome crystals in the presence of Mn2+ revealed the positions of metal ion cofactors within the assembled active site, confirming the predicted two-metal binding mode of retroviral INs (Fig. 1).
Intasome structure refinement using diffraction data collected on crystals soaked in the presence of HIV IN inhibitors Raltegravir or Elvitegravir revealed strong densities in the difference Fourier maps. The structures containing Raltegravir and Elvitegravir with pairs of metal atoms could be refined to 2.85 and 3.15 Å resolution, respectively (Fig. 1). Based on the structures, the drugs appear to have very similar modes of binding and action, involving an induced fit mechanism. Their metal chelating oxygen atoms interact with the metal cofactors of the active site, while their halobenzyl groups fit within a tight pocket created by dislocation of the 3′ adenosine. Within it, the drugs make intimate contacts to the bases of the invariant CA dinucleotide and conserved IN residues. In addition, the isopropyl and methyl-oxadiazole groups of Raltegravir are involved in hydrophobic and stacking interactions with conserved Pro-214 and Tyr-212, respectively (Fig. 1), further stabilizing the drug in the active site. Crucially, this mode of drug binding displaces the reactive 3′ viral DNA end from the active site, which can only result in deactivation of the intasome. Upon binding of Raltegravir, the reactive 3′ hydroxyl group moves away from the active site by more than 6 Å, compared to its positions in drug-free structures.
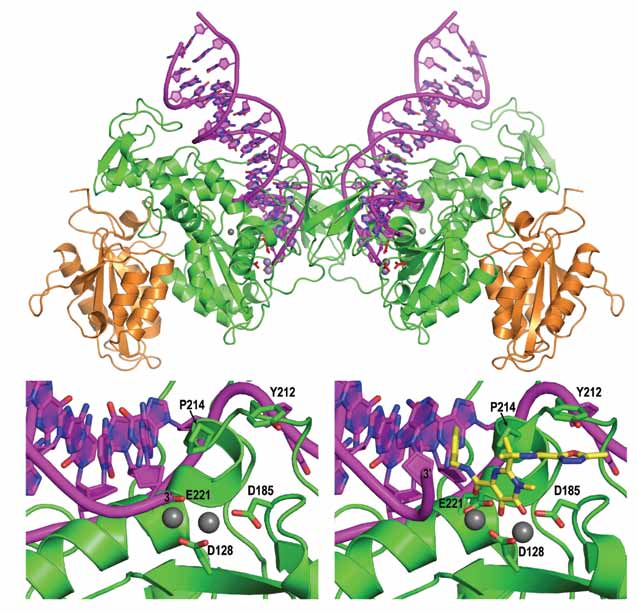 |
| Figure 1. Top panel: Overall organization of the retroviral intasome. Bottom: view on the active site in active (left) and Raltegravir-bound (right) states. Protein chains are shown as cartoons in green and orange, viral DNA in pink. Selected amino acid residues and the drug molecule (yellow) are shown as sticks; grey spheres are metal cations. |
Because the core contact points consisting of invariant nucleotide bases and amino acid residues are conserved, the mode of drug binding and action are unlikely to significantly differ between HIV-1 and PFV INs. The extensive contacts with the viral DNA end observed in our structures elucidate why the drugs preferentially interact with and inhibit the DNA-bound form of HIV-1 IN [5]. Furthermore, mutations of HIV-1 IN residue Tyr-143 (structural equivalent of PFV Tyr-212), which, based on our structure, is expected to interact with the methyloxadiazole group of Raltegravir (Fig. 1), are known to confer resistance to this drug [6]. Thus, PFV intasome structures allow building reliable models for HIV-1 IN and provide the long-needed platform for IN inhibitor design. Ongoing work inour group focuses on elucidation of the entire retroviral DNA integration pathway in snapshots of its key intermediates as well as mechanisms of HIV-1 resistance to strand transfer inhibitors.
References
[1] Hazuda DJ, Felock P, et al. Science, 287, 646-650 (2000).
[2] Summa V, Petrocchi A, et al. J Med Chem, 51, 5843-5855(2008).
[3] Jaskolski M, Alexandratos JN, et al. FEBS J, 276, 2926-2946. (2009)
[4] Valkov E, Gupta SS, et al. Nucleic Acids Res, 37, 243-255(2009).
[5] Grobler JA, Stillmock K, et al. Proc Natl Acad Sci USA, 99,6661-6666 (2002).
[6] Buzon MJ, Dalmau J, et al. AIDS, 24, 17-25 (2010).
Principal Publication and Authors
Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P.Retroviral intasome assembly and inhibition of DNA strand transfer, Nature, 464, 232-236 (2010).
Funding Acknowledgement
Medical Research Council, UK and National Institutes of Health, USA.

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.