
Find out more about our ambitious upgrade project, delivering more brightness, more coherence, and greater speed of analysis to UK science. More about Diamond-II

Find out more about Diamond's response to virus research.
Scientists have utilised Diamond Light Source to develop a new method to extract previously hidden information from the X-ray diffraction data that are measured when resolving the three-dimensional (3D) atomic structures of proteins and other biological molecules.
When trying to evolve chemical compounds towards potent drug candidates, scientists attempt to study the atomic detail of how compounds bind to their target proteins. To do so, they compare X ray data measured in both presence and absence of the compound. However, with existing analysis algorithms, this difference signal can often be swamped by noise from experiment artefacts, making it very unreliable to interpret the observed signal.
The new Pan-Dataset Density Analysis (PanDDA) method extracts the picture of the bound compound in exceptionally clear and unambiguous detail. PanDDA first identifies the source of the noise, and then removes it from the data. It exploits Diamond’s ability to repeat dozens to hundreds of measurements quickly, which are then characterised for differences between them, indicating the presence of bound compound, after which a noise correction is applied in 3D. The results are published today in Nature Communications.
Macromolecular crystallography (MX), the technique that PanDDA applies to, is one of the most powerful tools used by researchers interested in determining the 3D structures of large biological molecules, including proteins, and is the work-horse experiment for rational drug design.
“The problem of identifying binding events in crystallographic datasets can feel like looking for a needle in a haystack,” explains Dr Nicholas Pearce, lead author on the paper which comes from his PhD project at the University of Oxford in the Systems Approaches for Biomedical Science (SABS) Centre for Doctoral Training, where he was jointly funded by UCB Pharma and Diamond. “In the case of the data we were analysing, it was even worse, because we had hundreds of haystacks, and didn’t know which of them contained needles.” Dr Pearce is now based in the Crystal & Structural Chemistry Group at Universiteit Utrecht.
The researchers were able to use to their advantage the fact that most of the measurements were from ‘empty’ crystals that didn’t contain a bound ligand, allowing them to characterise the unbound form and simply looking for datasets that were different.
“Often in crystallography you can miss ‘weak’ bound forms, because each measurement is a superposition of the bound and unbound forms,” continues Dr Pearce. “This is akin to multiple sheets of tracing paper, each with one of at least two images, all overlaid on top of each other.”
“When trying to identify the image on only one of the ‘sheets’, it gets confused by what shows through from all the other sheets, so the image becomes susceptible to interpretation errors,” Dr Pearce adds. “To overcome this, we developed a method to extract the right set of ‘sheets’ from the superposition; once we’d done that, interpreting the bound form becomes much easier, and enables us to confidently interpret the data, and build models of the interesting states in the data.”
“The basic idea is conceptually very simple, namely treating the confusing superposition as a background correction problem,” explains Professor Frank von Delft, who is jointly Principal Investigator of the Protein Crystallography group in the Structural Genomics Consortium (SGC) at the University of Oxford, and Principal Beamline Scientist of the I04-1 beamline at Diamond. “However, an accurate estimate of the background is crucial, and in practice this was unthinkable until the advent of the new robotic technology offered by Diamond, which makes it routine to make such large numbers of measurements.”
“UCB is delighted to have been working closely with Diamond on the development of PanDDA and its application to crystallographic fragment screening,” comments Dr Neil Weir, Senior Vice President of Discovery at UCB Pharma. “As a direct result, we have been able to identify fragments, which were otherwise not distinguishable from background, bound to a key protein-protein interaction drug target.”
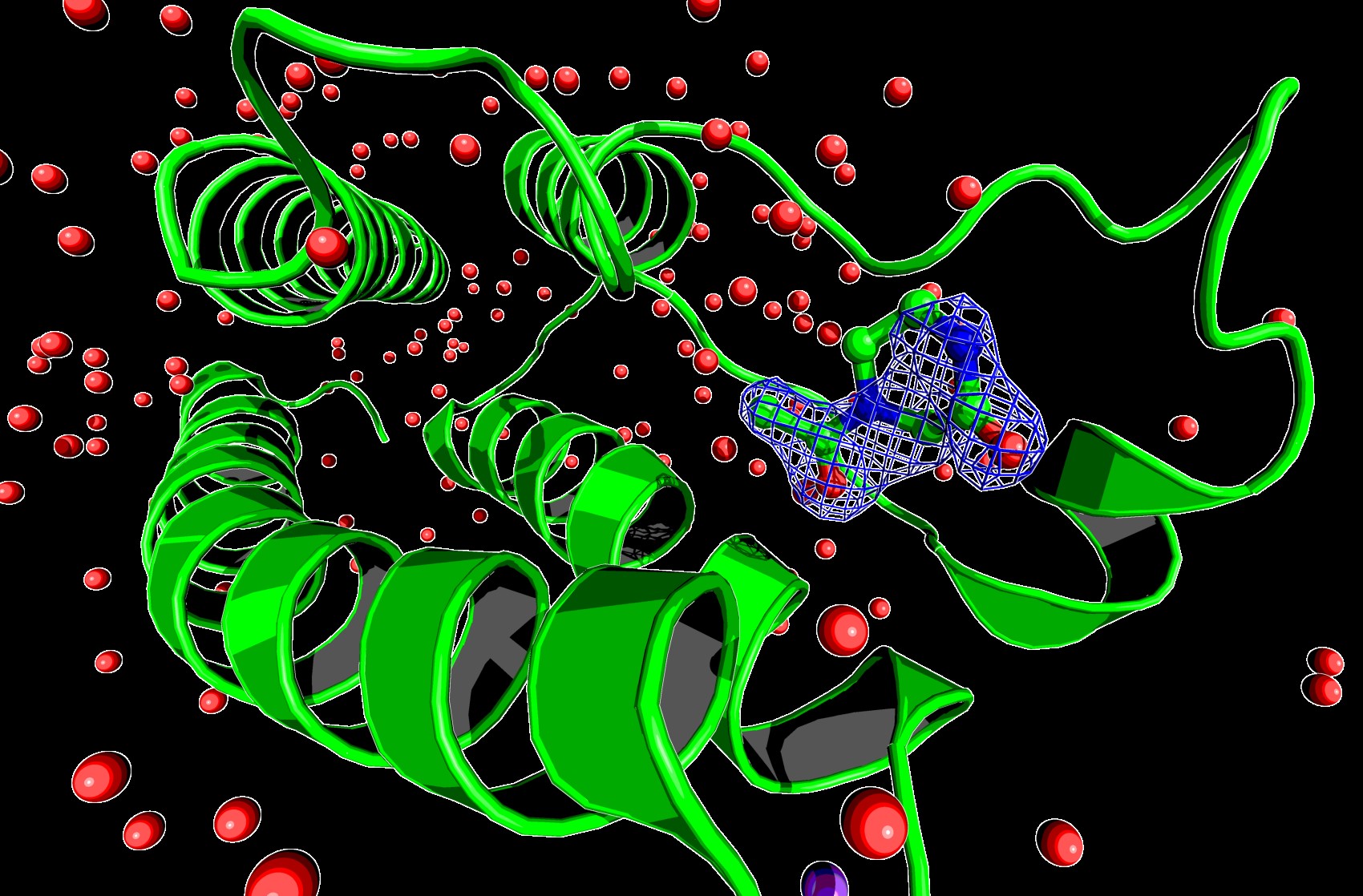
The research involved producing around 860 datasets, of which only 75 contain a bound form of interest to the researchers. “While general applicable in MX, the method is particularly transformative for a version of the MX experiment called fragments screening, where the effects we’re looking for are very rare and even harder to verify by conventional algorithms,” continues von Delft.
A crucial coda to the work was the uploading of all the structures to the Protein Data Bank (wwPDB), the online repository of 3D structures of proteins and nucleic acids, where everybody has completely free access to all structures ever published. One of the wwPDB host sites, RCSB PDB, recently developed a new group deposition tool to allow the mass upload of structures, and this was crucial to completing this collaboration.
The RCSB PDB Group Deposition system allows authors to take advantage of local templates and PDB_extract for batch processing, packing, upload, review, validation, and one-click submission of many structures at once. Searching group title “PanDDA analysis group deposition” at rcsb.org will return these 860 depositions.
“The Diamond and PDB groups have accomplished something quite incredible, and we have been delighted to help them” says Aled Edwards, Director of the SGC. “I would also like to highlight the team’s commitment to open science. By placing all the research output into the public domain, they have ensured that the data can be used by all.”
Now celebrating its 10th year of research and innovation, Diamond is committed to working with our users to enable them to carry out world-leading research at the facility.
“We’ve come a long way in the last ten years, and collaborations like these are key to how we will maintain our place as a key facility for researchers working in the life sciences,” adds Professor Dave Stuart, Director of Life Sciences at Diamond. “The idea that we can clearly see weak binding events is particularly exciting and something we’re looking forward to sharing with our crystallography community.”
The researchers hope that this new method will provide a significant shift in how crystallographic models are generated; opening windows to explore more poorly ordered crystals.
Pearce, N. M. et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 8, 15123 doi: 10.1038/ncomms15123 (2017), is available at: www.nature.com/articles/ncomms15123.

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Copyright © 2022 Diamond Light Source
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.

 A brighter light for science
A brighter light for science