 | ||
AP2 structure illuminates regulation of clathrin-mediated endocytosis |
The cell’s external membrane is an impermeable barrier, made selectively permeable to some molecules (such as nutrients or chemical signals) by membrane-spanning proteins such as ion channels and receptors. The complement of these proteins at the cell surface is tightly controlled by continuous removal and replenishment. Clathrin-mediated endocytosis (CME) is a key mechanism that accomplishes this task. In CME, receptors bound to adaptor proteins such as AP2 are packaged into vesicles with the aid of a clathrin coat to form clathrin-coated vesicles (CCVs). It is fundamental to many cell surface functions such as neurotransmission and signal transduction and is thus essential to cell life.
The formation of CCVs occurs due to specialised proteins called ‘clathrin adaptor proteins’ that attract the clathrin molecules and also decide what the CCVs are going to transport from outside to inside the cell. Using Diamond Light Source's Macromolecular Crystallography (MX) beamlines I02, I03, and I04-1, a key clathrin adaptor protein (AP2) was studied to see how it recruited and polymerised clathrin and what switches this process on and off. The results obtained revealed that the part of AP2, a clathrin binding motif, that gathers and forms the clathrin into CCVs was ‘locked’ or inactive. Furthermore, the transition of this motif to the active or ‘open’ conformation was triggered by binding of the AP2 to certain areas of the plasma membrane. These results provide us with important structural information to better understand this vital cell process
Beamlines I02/I03/I04-1 Scientific Highlight
The central 200 kDa core of AP2 comprises the N-terminal alpha-helical solenoid domains of the α and β2 subunits, together with the μ2 and σ2 subunits (Fig. 1a,b). The α and β2 subunits further possess flexible ‘hinge’ and C-terminal ‘appendage’ domains (Fig. 1b). Previous work1 showed that the core exists in two different conformations: a ‘locked’ or inactive conformation, incapable of binding endocytic cargo due to blockage of the binding sites, and an ‘open’ membrane-interacting and ligand-bound form. This suggested a model in which cytosolic AP2 is prevented from inappropriate recognition of the cargo motifs (Yxx[ILVMF] and [ED]xxxL[LI]) that mark a transmembrane protein as endocytic cargo, but that are also found in cytosolic (i.e. non-cargo) proteins. Recruitment of AP2 to the plasma membrane, driven by binding of AP2 to the plasma membrane-specific phosphoinositide, PtdIns(4,5)P2, stimulates the dramatic conformational change to the open form, which is capable of binding endocytic cargo and so should be incorporated into forming endocytic CCVs.
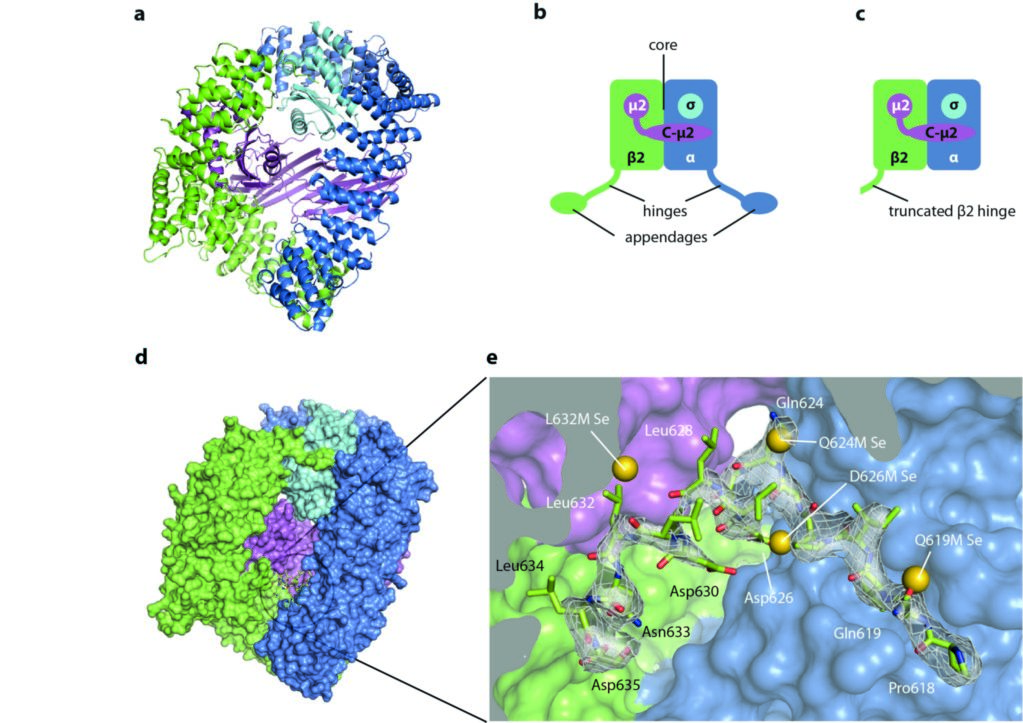
Figure 1: Structure of AP2 and sequestration of the LLNLD clathrin-binding motif (residues 631-635 of β2); (a) the AP2 core in cartoon representation; (b) schematic of the whole AP2 complex, showing the ‘hinges’ and ‘appendages’ of α and β2 in addition to the core; (c) the extended core construct crystallised in this study, lacking the α and β appendages and the α hinge; (d) surface representation of the extended core structure; (e) close up view of the buried β2 hinge residues. The selenium sites located in four of the methionine point mutants are shown as gold spheres. Adapted from Science 2014 Jul 25;345(6195):459-63.
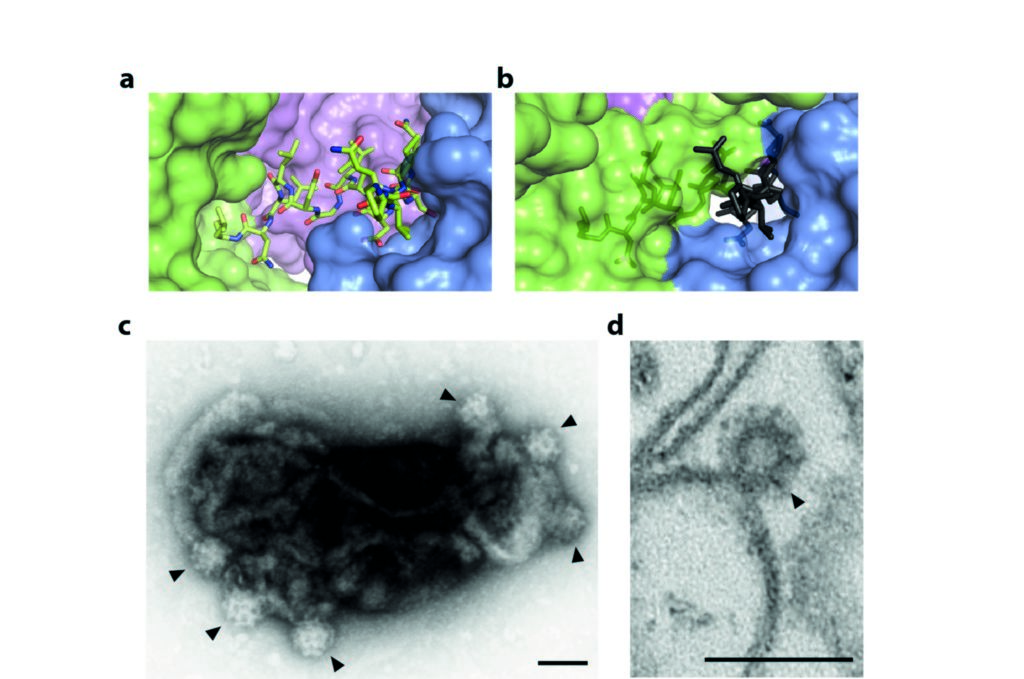
Figure 2: The mechanism of regulated clathrin recruitment in AP2; (a) the buried part of the β2 hinge in the locked form of AP2; (b) the same fragment of β2 superposed in grey on the open form of AP2, aligned with the locked form on the part of the α subunit abutting the binding site, showing that burial of the LLNLD clathrin-binding motif is incompatible with this conformation of AP2; (c) clathrin-coated ‘bud’ formation (indicated by arrowheads) on synthetic liposomes containing PtdIns(4,5)P2 and lipid-linked cargo peptide visualised by negative-stain EM and (d) by ultrathin sectioning. Scalebars are 100 nm. Adapted from Science 2014 Jul 25;345(6195):459-63.
Using custom scripts, a data processing pipeline was assembled, taking advantage of xia22 to automate integration and scaling, and PHASER-EP3 to find selenium sites. The latter performs iterative substructure completion using log-likelihood gradient maps, a sensitive technique for the robust location of weak anomalous scatterer sites. This analysis revealed the presence of selenium sites at all the methionine residues of the AP2 core, but also at positions corresponding to the buried part of the hinge in four of the mutants tested. The positions of these hinge selenium sites were consistent with a model in which residues abutting the clathrin-interaction motif are buried (Fig. 1e).
These data suggest a model that explains the observed PtdIns(4,5)P2- and cargo-stimulated recruitment of clathrin. In the locked, cytosolic state, clathrin binding by AP2 is inhibited because the main clathrin-interaction site in the hinge of β2 is buried and inaccessible to clathrin (Fig. 2a). Upon recruitment to the plasma membrane via PtdIns(4,5)P2, AP2 undergoes a dramatic conformational change that is stabilised by binding to endocytic cargo. In this open structure (PDB ID 2XA7), the entrance to the deep hinge-binding cleft is blocked because the α and β2 subunits swing towards each other (Fig. 2b). Thus, in transitioning from the locked to the open conformation, the β2 hinge must be expelled from its binding site, allowing clathrin recruitment. Negative stain electron microscopy experiments showed that recombinant AP2 with a full-length β2 subunit was able to recruit and polymerise clathrin on synthetic liposome membranes containing PtdIns(4,5)P2 and endocytic cargo motif peptide, forming proto-transport vesicles termed ‘buds’ (Fig. 2c,d). Together, these results suggest a model in which AP2 is prevented from driving clathrin polymerisation except at sites of endocytic transport vesicle formation.
Source publication:
Kelly, B. T., Graham, S. C., Liska, N., Dannhauser, P. N., Hoening, S., Ungewickell, E. J. & Owen, D. J. AP2 controls clathrin polymerization with a membrane-activated switch. Science 345, 459-463, doi:10.1126/science.1254836 (2014).
References:
1. Jackson, L. P. et al. A Large-Scale Conformational Change Couples Membrane Recruitment to Cargo Binding in the AP2 Clathrin Adaptor Complex. Cell 141, 1220-U1213, doi:10.1016/j.cell.2010.05.006 (2010).
2. Winter, G. xia2: an expert system for macromolecular crystallography data reduction. Journal of Applied Crystallography 43, 186-190, doi:10.1107/ s0021889809045701 (2010).
3. Read, R. J. & McCoy, A. J. Using SAD data in Phaser. Acta Crystallographica Section D-Biological Crystallography 67, 338-344, doi:10.1107/ s0907444910051371 (2011).
Funding acknowledgement:
Wellcome Trust Principal Research Fellowship (to DJO), award number 090909.
Corresponding authors:
Dr Bernard Kelly, Cambridge Institute for Medical Research, [email protected]; Professor David Owen, Cambridge Institute for Medical Research, [email protected]
