Toward remote control of an ‘undruggable’ dynamic enzyme with structurebased fragment screening
Jun 27, 2019
Jun 27, 2019
Many treatments for human diseases use small-molecule drugs that target protein molecules. It is sometimes quite difficult to target the active site within the protein (or ‘business end’) with such molecules. An international team of researchers set out to find new sites on a particular medically promising protein - PTP1B - where drug-like small molecules might bind and alter how the protein behaves, with the longer-term goal of treating diseases such as diabetes and breast cancer.
Their goal was to expose the protein of interest, called PTP1B, to nearly 2,000 small molecules, and to use X-ray crystallography to learn which parts of PTP1B are most prone to having molecules bind to them. They used the automated XChem fragment screening facility on beamline I04-1, followed by computer-based analysis to process the large amount of data collected during this work.
Proteins are dynamic molecules whose motions are important for their biological functions. In allostery, for example, protein motions between alternative conformations can propagate a signal from one part of a protein structure to another part where it can affect structure, dynamics, and function. Because protein energy landscapes are complex and inevitably give rise to alternative conformations, allostery is increasingly thought to be a property of potentially all proteins1. However, allostery remains incompletely understood, in part because we lack structural biology approaches that allow us to interrogate how different parts of a protein structure are ‘wired’ together, and which surface sites can act as inputs to this wiring network by binding small-molecule allosteric modulators.
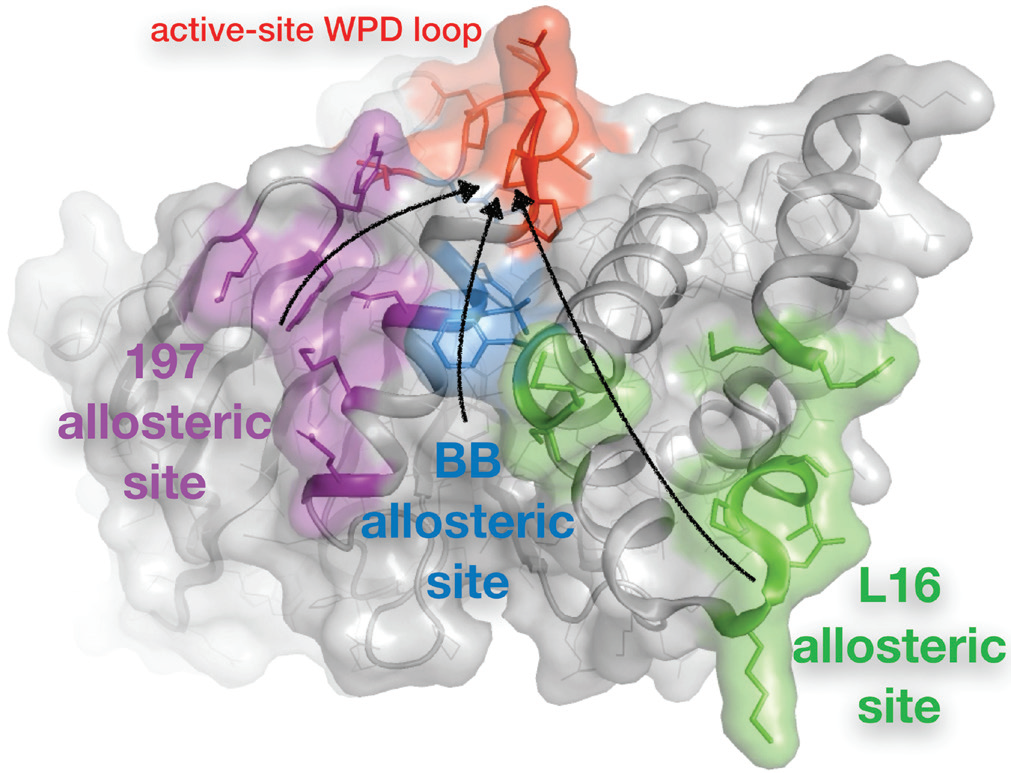
The first part of this work, performed separate from Diamond, sought to elucidate the allosteric network inherent to unliganded PTP1B. To do so, multitemperature crystallography4 – instead of the more traditional cryogenic-temperature crystallography – was used to reveal redistributions of electron density for particular residues as a function of temperature. Multiconformer modelling algorithms were then used to model the corresponding alternative conformations in atomic detail. Connecting the dots between the interacting, flexible residues in PTP1B revealed a substantial putative allosteric network that included three main allosteric sites (Fig. 1). One of these sites, the BB site, coincides with the binding site for the previously reported but abandoned allosteric inhibitor3. The other sites had not been identified previously, though similar regions were implicated by concurrent analyses based on Nuclear Magnetic Resonance (NMR) spectroscopy and mutagenesis.
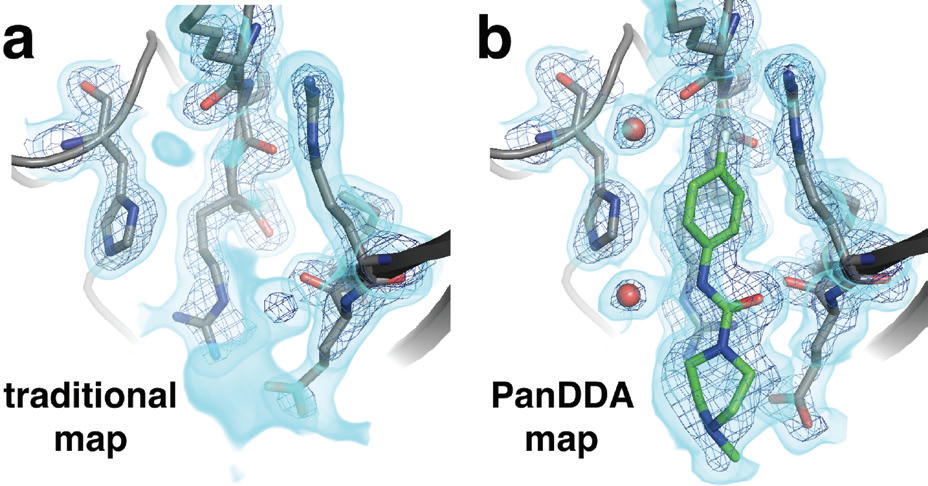
Importantly, by virtue of their small size, small-molecule fragments typically bind at low occupancy, and are therefore often undetectable using traditional 2Fo-Fc electron density maps (Fig. 2a). However, this work took advantage of the recently developed Pan-Dataset Density Analysis (PanDDA) algorithm5, which exploits large numbers of datasets, like those collected at XChem, to compute an average ‘background’ model for the unbound state and computationally subtract it, thus yielding a clear view of the low-occupancy bound state. For our PTP1B datasets, PanDDA yielded excellent density maps for low-occupancy fragment binding events (Fig. 2b) scattered across the protein surface; these binding events would not have been evident without this special analysis.
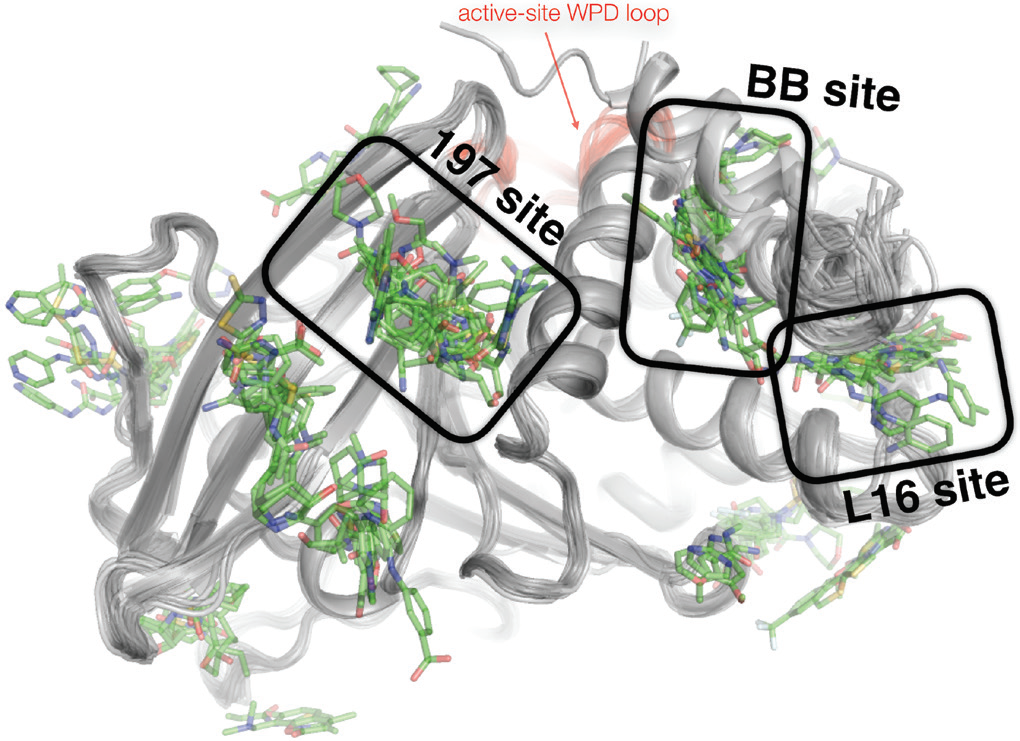
Overall, most fragments did not bind, as was expected given the naively constructed fragment library. However, using PanDDA, 110 of the 1,774 datasets yielded all-atom structures of PTP1B bound to unique small-molecule fragments. These fragments’ binding sites were spread across the surface of PTP1B – but they clustered at three primary ‘hot spots’ (Fig. 3) which coincided remarkably well with the putative allosteric sites discovered by multitemperature crystallography of the apo enzyme (Fig. 1). This convergence was encouraging that these sites in PTP1B would perhaps be capable of binding potent allosteric inhibitors.
To separately validate the functional relevance of the putative allosteric sites identified by XChem fragment screening of PTP1B, a covalent tethering experiment was also performed outside of Diamond. One molecule, compound 2, significantly inhibited PTP1B catalysis (Ki of 7.1 ± 1.1 μM) when tethered to a K197C mutation at the new 197 allosteric site, confirming that at least this site is truly allosterically linked to the catalytic site.
References:

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Copyright © Diamond Light Source. Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.