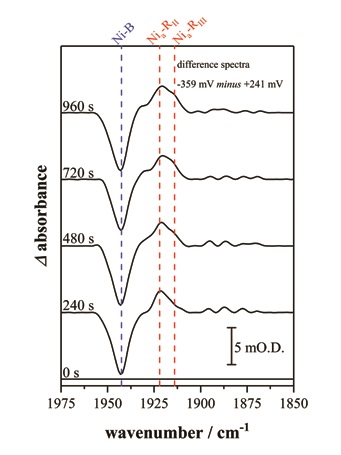
- Figure 2: This sequence of infrared spectra highlight the time dependent formation of two of the Nia–R sub-states, Nia–RII and Nia–RIII in hydrogenase I from Escherichia coli. Spectra are presented as reduced minus oxidised baseline-corrected difference spectra following application of a reducing potential step from +241 to -359 mV (vs the standard hydrogen electrode, SHE)
This study exploited the synchrotron IR source of the Multimode IR Imaging and Microspectroscopy (MIRIAM) beamline, Diamond Light Source, to monitor the active site of [NiFe] hydrogenase 1 from E. coli in the crystalline state as a case study5. Hydrogenase 1 yields large well-diffracting crystals, with a typical cross-section of 50 × 50 μm2 and axial length of 1000 – 2000 μm. A custom-built reflection-absorption three-electrode electrochemical cell allowed precise potential control, with a cocktail of redox mediators in solution employed to facilitate electron transfer between the crystal and working electrode. Spectra recorded following alternating reductive and oxidative potential steps confirmed that electrochemical control of the hydrogenase 1 active site was fully reversible over multiple reduction-oxidation sequences. The brightness of the MIRIAM source enabled collection of high signal-to-noise spectra at discrete 15 × 15 μm2 sub-sections along the length of single hydrogenase 1 crystals, and electrochemical control was shown to extend over the whole crystal length. Spectral changes were complete within 30 minutes of application of a potential step.
The [NiFe] hydrogenase catalytic cycle (Fig. 1b) consists of both electrochemical steps and chemical steps involving proton transfer. The most reduced equilibrium state of the [NiFe] hydrogenase active site, Nia-R, actually consists of three sub-states at the same redox level. The precise nature of the Nia-R sub-states is unknown but they are thought to represent differently protonated forms of the [NiFe] active site, possibly indicative of discrete proton transfer events following initial activation of H22. In solution and steady-state turnover studies the different Nia-R sub-states states are observed at constant ratio, regardless of the potential, presumably interchanging on a timescale faster than the spectroscopic measurement. Interestingly in the crystalline state the interconversion of individual Nia-R sub-states can be temporally resolved, providing the first evidence that these sub-states might be sequential intermediates in the catalytic cycle (Fig. 2).
The results of this study have two important implications. Firstly, the ability to control, electrochemically, the redox state of a metalloprotein in a single protein crystal offers possibilities for obtaining ‘snapshots’ of complex redox proteins in well-defined states that have not been accessible previously. When combined with imaging of crystals using IR microspectroscopy, the state of the protein in the crystal can be checked before X-ray crystallography is used to obtain a structural ‘snapshot’, and afterwards to check whether there has been any damage to the crystal during the structural measurement. Secondly, our observation that chemistry is slowed down inside the hydrogenase crystal offers possibilities of trapping out specific states that react too fast to be detected in alternative solution studies. Thus, this body of work represents an important advance for understanding the interplay between structure and function in some of the complex metal-containing proteins that are critical to life.
![Figure 1: (a) Structure of the [NiFe] hydrogenase I from Escherichia coli in cartoon form, with the metal centres and their ligands shown as spheres in elemental colours (nickel: green, iron: orange, sulphur: yellow, carbon: grey, oxygen: red, nitrogen: blue); (b) Proposed catalytic cycle at the active site of [NiFe] hydrogenases.](/dam/jcr:b53ef53c-be68-4d7e-b9ef-68cbeab4ec8a/p85fig1.jpg)
