 | ||
Controlling the warp and weft of molecules that are knitted together |
Related publication:
Wood CS, Ronson TK, Belenguer AM, Holstein JJ, Nitschke JR. Two-stage directed self-assembly of a cyclic [3] catenane. Nature Chemistry 7, 354-358, doi:10.1038/nchem.2205 (2015).
Keywords:
Self-assembly; Coordination chemistry; Catenation.
The Small Molecule Single Crystal Diffraction beamline (I19) has helped elucidate a novel interlocked molecular structure made up of three interlinked rings, like a nanoscale version of chainmail. Such interlocked molecular structures could lead to materials with novel properties, but they have proved difficult to produce because of a paucity of methods for weaving or knotting multiple chains of atoms together so they can’t unravel or unlink. Producing the new interlinked ring structure, termed a cyclic [3]catenane, thus required the development of a completely new synthesis method.
This new method used a metal-templated self-assembly procedure involving five distinct precursor molecules to generate the rings, with each ring designed to connect with two other rings. Developing this method required structural information provided by I19, with this information comprising the crystal structures of both the precursor framework and the final interlinked ring structure. By revealing how the rings of atoms were positioned within a molecule and how to connect them selectively, the structural information was critical for ensuring that the precursor molecules and rings linked together in the correct way. This metal-templated self-assembly method could now be adapted to produce other interlocked molecular structures, including larger and more complex structures.From the interlocked rings of chainmail to the knots holding climbers to cliff faces, interlacing linear strands into precisely defined three-dimensional patterns has allowed the creation of new devices and materials throughout history. A current challenge for chemists is applying the principles of knotting and interlocking at the nanometre scale and below. Key to achieving this is to obtain the knowledge of exactly how strands or rings of atoms lie within a molecule and how to connect them selectively. The related publication reports a new method for the formation of such mechanically interlocked molecules developed with the aid of structural information obtained from experiments on beamline I19.
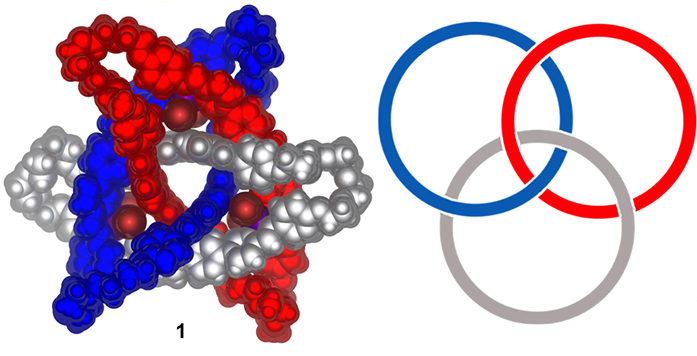
Figure 1: a) Space-filling representation of the crystal structure of metal-organic cyclic [3]catenane 1 with non-coordinating anions, solvent and disorder omitted for clarity; b) representation of the topology of a cyclic [3]catenane
The paper by Wood and colleagues describes the synthesis of a precisely ordered circular double helicate, and its mechanical interlocking to form a molecule consisting of three interwoven rings (Fig. 1). The method developed retains the intricate geometry of the original helicates to create a topologically chiral cyclic [3]catenane structure.
The precise interleaving of the different ligands in the precursor double triangle 4 was achieved by exploiting the principle of coordinative saturation1 in a self-assembling system with several different ligands. Ligand 2 (Fig. 2) was designed to disfavour the formation of homoleptic (single-ligand) selfassembled structures in the presence of iron(II) cations and thus preferentially form heteroleptic (mixed-ligand) structures in reaction mixtures where other types of ligand are present.
Combinations of ligands 2 and 3 with iron(II) cations and bromide anions in acetonitrile led to the self-assembly of a mixed ligand circular helicate 4 (Fig. 2). Data obtained in experiments using beamline I19 were critical in elucidating the structure, which was shown to comprise two interwoven trinuclear circular helicates, linked by coordination of the three innermost iron(II) centres. The remaining three outermost iron(II) centres were found to feature coordination sites not satisfied by either ligands 2 or 3 and were instead coordinated by bromide.
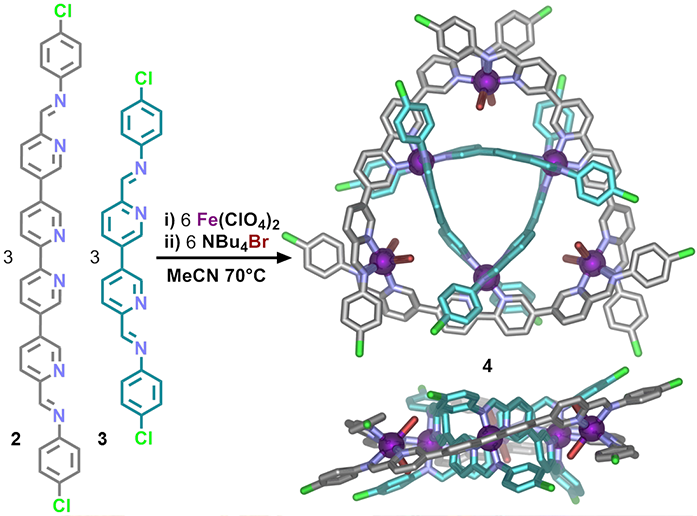
Figure 2: Synthesis and X-ray crystal structure of mixed-ligand circular double helicate 4. Hydrogen atoms, disorder, non-coordinated anions and solvent are omitted for clarity (C: grey or turquoise, N: blue, O: red, Br: brown, Fe: purple, Cl: green).
The low ligand field strength of Br- stabilised the high-spin state of these iron(II) centres, causing the complex to exhibit paramagnetism, as observed in the 1H NMR. Confirmation of the high-spin state of the outer iron(II) centres was obtained from studying the Fe-N bond lengths in the crystal structure. The N-Fe bond lengths for the outer iron(II) centres were observed to be in the range of 2.147(4)-2.304(5) Å which is significantly longer than the 1.960(4)- 1.997(4) Å observed for the low-spin inner iron(II) centres.
The proximity and orientation of the chloroaniline residues that make up the pyridylimine coordination sites of ligands 2 and 3 in structure 4 led to the hypothesis that sites occupied by pairs of these residues could be selectively linked by using a suitable diamine. Although one could envisage linking different potential pyridylimine sites within 4, it was hypothesised that control over diamine length and geometry would permit the two closest metalcoordinated imine sites on the same face to be selectively linked.
Syntheses using a range of flexible and rigid diamines using a one-pot synthesis method, whereby all the building blocks were combined directly, led to a range of insoluble precipitates. To overcome this problem, subcomponent substitution2, whereby electron-rich anilines displace electron-poor aniline residues, was employed to achieve the interlocking.
Metal-organic cyclic [3]catenane 1 was synthesised via the selective substitution of the twelve 4-chloroanline residues within preformed 4 by six electron-rich dianilines that incorporated flexible linking chains (Fig. 3). Here, once again, data obtained from single crystal X-ray experiments provided critical information as to the precise location of the introduced dianlines and thus the new molecule’s interlocked nature.
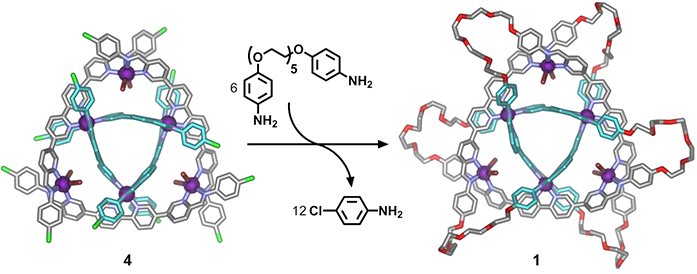
Figure 3: Subcomponent substitution of 4 with an electron rich dianiline yielded cyclic [3]catenane 1, shown as the crystal structure. Hydrogen atoms, disorder, non-coordinated anions and solvent are omitted for clarity (C: grey or turquoise, N: blue, O: red, Br: brown, Fe: purple, Cl: green).
Further evidence for the interlocked nature of 4 was provided by LC-MS studies on a wholly organic cyclic [3]catenane generated by the sodium borohydride mediated reduction of 1. The reduction generated a mixture of the reduced and demetallated [3]catenane, the related [2]catenane, and the macrocycle formed via the hydrolytic dethreading of the rings3. MS/ MS studies on the cyclic [3]catenane species showed that by breaking the rings via energetic gas bombardment in the gas phase generated a species corresponding to a [2]catenane product. These results are consistent with the ring connectivity of a cyclic [3]catenene, whereby if one ring breaks, the remaining rings stay locked together.
Although practical applications are not the focus of this study, the precise control over molecular interlinkages may enable the generation of new materials with different properties that are otherwise difficult to combine – flexibility and tensile strength, for example.
References:
- Kramer, R., Lehn, J. M. & Marquis-Rigault, A. Self-recognition in helicate self-assembly: spontaneous formation of helical metal complexes from mixtures of ligands and metal ions. Proceedings of the National Academy of Sciences of the United States of America 90, 5394-5398 (1993).
- Schultz, D. & Nitschke, J. R. Designing multistep transformations using the Hammett equation: Imine exchange on a copper(I) template. Journal of the American Chemical Society 128, 9887-9892, doi:10.1021/ja061841u (2006).
- Peters, A. J., Chichak, K. S., Cantrill, S. J. & Stoddart, J. F. Nanoscale Borromean links for real. Chemical Communications 3394-3396, doi:10.1039/b505730b (2005).
Funding acknowledgement:
This work was supported by the UK Engineering and Physical Sciences Research Council (EPSRC, EP/I005447/1) and a Marie Curie fellowship for JJH (ITN-2010-264645). We thank Diamond Light Source (UK) for synchrotron beamtime on I19 (MT7984 and MT8464).
Corresponding authors:
Christopher S. Wood, Professor Jonathan R Nitschke, University of Cambridge, [email protected]
