
___________________________________
Industrial Liaison Group:
Tel: +44 (0) 1235 778797
E-mail: [email protected]
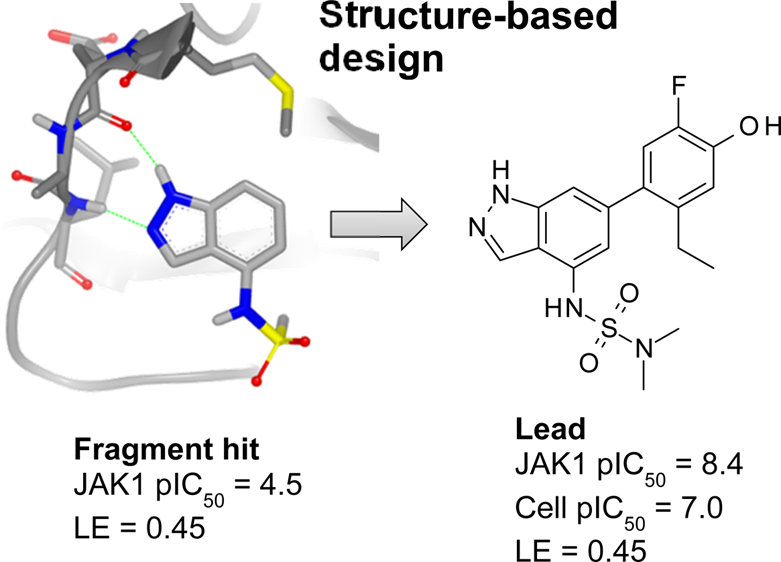
Abstract
Janus kinase (JAK) inhibitors are emerging as novel and efficacious drugs for treating psoriasis and other inflammatory skin disorders, but their full potential is hampered by systemic side effects. To overcome this limitation, we set out to discover soft drug JAK inhibitors for topical use. A fragment screen yielded an indazole hit that was elaborated into a potent JAK inhibitor using structure-based design. Growing the fragment by installing a phenol moiety in the 6-position afforded a greatly improved potency. Fine-tuning the substituents on the phenol and sulfonamide moieties afforded a set of compounds with lead-like properties, but they were found to be phototoxic and unstable in the presence of light.
ACS Med. Chem. Lett.(2016), DOI: 10.1021/acsmedchemlett.6b00087
Andreas Ritzén*†, Morten D. Sørensen†, Kevin N. Dack†, Daniel R. Greve†, Anders Jerre‡, Martin A. Carnerup†, Klaus A. Rytved§, and Jesper Bagger-Bahnsen∥
†Drug Design, ‡In Vitro Biology, §Skin PK and Early Safety, and ∥Preformulation & Early Analytical Development, Global R&D, LEO Pharma A/S, Industriparken 55, DK-2750 Ballerup, Denmark
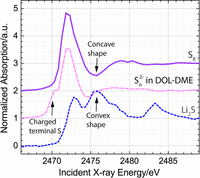
Abstract
Replacement of conventional cars with battery electric vehicles (BEVs) offers an opportunity to significantly reduce future carbon dioxide emissions. One possible way to facilitate widespread acceptance of BEVs is to replace the lithium-ion batteries used in existing BEVs with a lithium-sulfur battery, which operates using a cheap and abundant raw material with a high specific energy density. These significant theoretical advantages of lithium-sulfur batteries over the lithium-ion technology have generated a lot of interest in the system, but the development of practical prototypes, which could be successfully incorporated into BEVs, remains slow. To accelerate the development of improved lithium-sulfur batteries, our work focuses on the mechanistic understanding of the processes occurring inside the battery. In particular, we study the mechanism of the charging process and obtain spatially resolved information about both solution and solid phase intermediates in two locations of an operating Li2S-Li battery: the cathode and the separator. These measurements were made possible through the combination of a spectro-electrochemical cell developed in our laboratory and synchrotron based operando X-ray absorption spectroscopy measurements. Using the generated data, we identify a charging mechanism in a standard DOL-DME based electrolyte, which is consistent with both the first and subsequent charging processes.
J. Electrochem. Soc. 2016 volume 163, issue 6, A930-A939 doi: 10.1149/2.0631606jes
Yelena Gorlina,*,z Manu U. M. Patela, Anna Freiberga,*, Qi Hea, Michele Pianaa,**, Moniek Trompb and Hubert A. Gasteigera,***
aChair of Technical Electrochemistry, Department of Chemistry and Catalysis Research Center, Technische Universität München, Garching, Germany
bVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Amsterdam, Netherlands
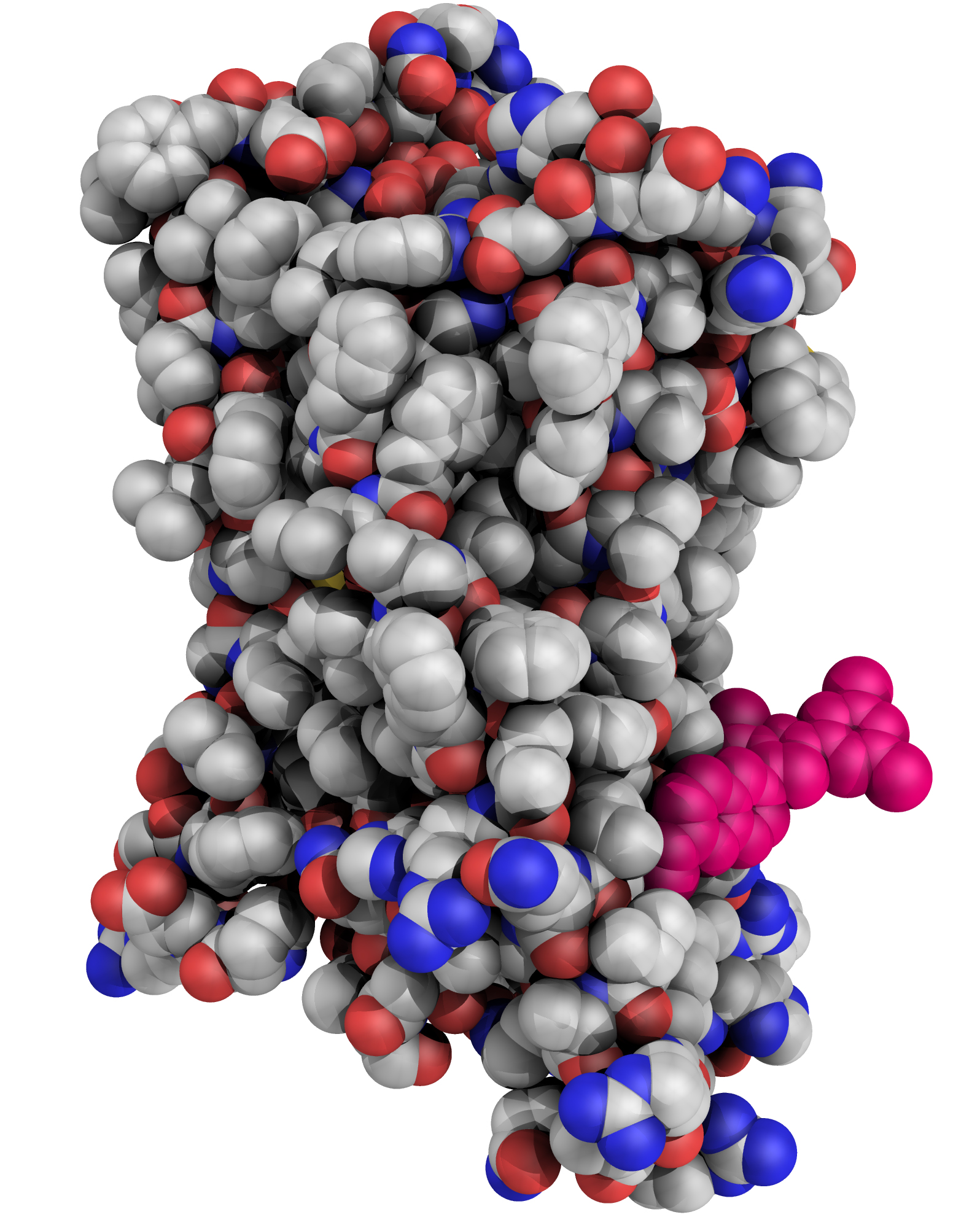
Abstract
Glucagon is a 29-amino-acid peptide released from the α-cells of the islet of Langerhans, which has a key role in glucose homeostasis1. Glucagon action is transduced by the class B G-protein-coupled glucagon receptor (GCGR), which is located on liver, kidney, intestinal smooth muscle, brain, adipose tissue, heart and pancreas cells, and this receptor has been considered an important drug target in the treatment of diabetes. Administration of recently identified small-molecule GCGR antagonists in patients with type 2 diabetes results in a substantial reduction of fasting and postprandial glucose concentrations2. Although an X-ray structure of the transmembrane domain of the GCGR3 has previously been solved, the ligand (NNC0640) was not resolved. Here we report the 2.5 Å structure of human GCGR in complex with the antagonist MK-0893 (ref. 4), which is found to bind to an allosteric site outside the seven transmembrane (7TM) helical bundle in a position between TM6 and TM7 extending into the lipid bilayer. Mutagenesis of key residues identified in the X-ray structure confirms their role in the binding of MK-0893 to the receptor. The unexpected position of the binding site for MK-0893, which is structurally similar to other GCGR antagonists, suggests that glucagon activation of the receptor is prevented by restriction of the outward helical movement of TM6 required for G-protein coupling. Structural knowledge of class B receptors is limited, with only one other ligand-binding site defined—for the corticotropin-releasing hormone receptor 1 (CRF1R)—which was located deep within the 7TM bundle5. We describe a completely novel allosteric binding site for class B receptors, providing an opportunity for structure-based drug design for this receptor class and furthering our understanding of the mechanisms of activation of these receptors.
Nature (2016) doi:10.1038/nature17414
Ali Jazayeri, Andrew S. Doré, Daniel Lamb, Harini Krishnamurthy, Stacey M. Southall, Asma H. Baig, Andrea Bortolato, Markus Koglin, Nathan J. Robertson, James C. Errey, Stephen P. Andrews, Iryna Teobald, Alastair J. H. Brown, Robert M. Cooke, Malcolm Weir & Fiona H. Marshall
Heptares Therapeutics Ltd, BioPark, Broadwater Road, Welwyn Garden City, Hertfordshire AL7 3AX, UK
Week commencing 25th April 2016
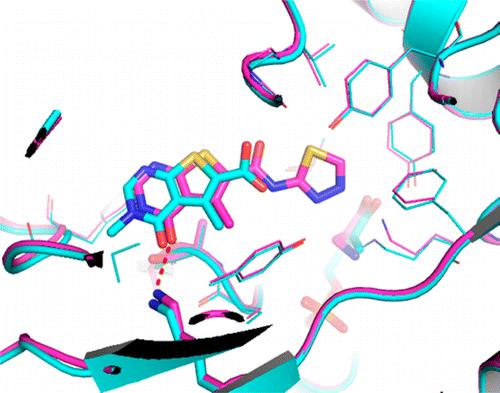
Abstract
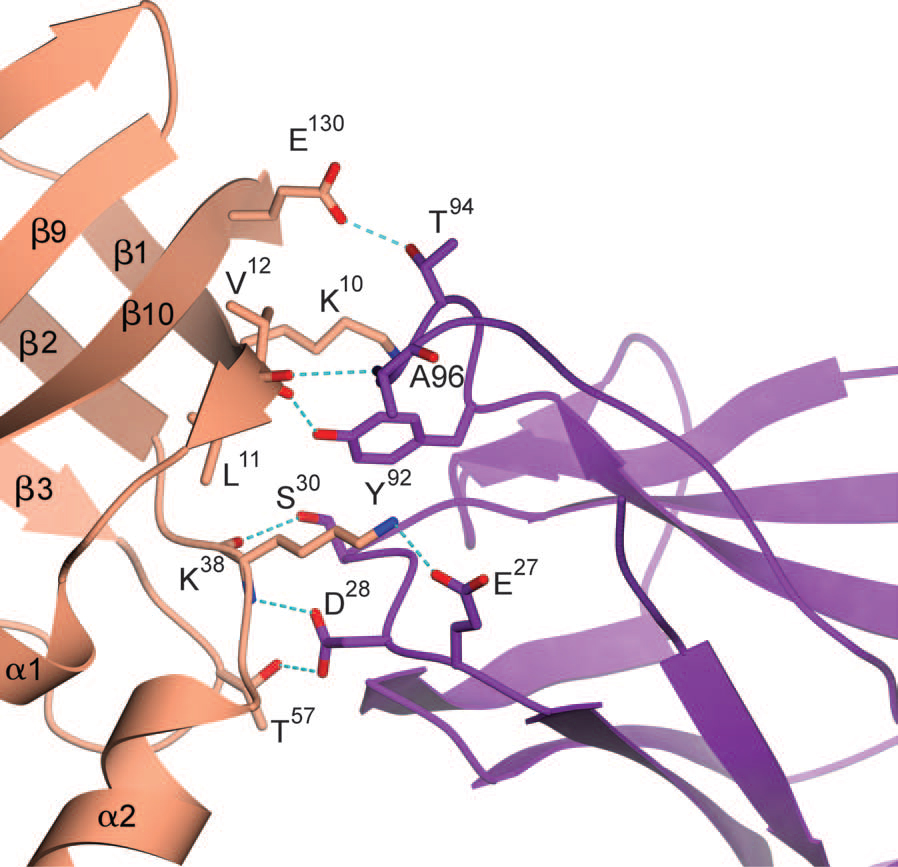
Inhibitors of mitochondrial branched chain aminotransferase (BCATm), identified using fragment screening, are described. This was carried out using a combination of STD-NMR, thermal melt (Tm), and biochemical assays to identify compounds that bound to BCATm, which were subsequently progressed to X-ray crystallography, where a number of exemplars showed significant diversity in their binding modes. The hits identified were supplemented by searching and screening of additional analogues, which enabled the gathering of further X-ray data where the original hits had not produced liganded structures. The fragment hits were optimized using structure-based design, with some transfer of information between series, which enabled the identification of ligand efficient lead molecules with micromolar levels of inhibition, cellular activity, and good solubility.
J. Med. Chem., 2016, 59 (6), pp 2452–2467 DOI: 10.1021/acs.jmedchem.5b01607
Jennifer A. Borthwick,*,†,§ Nicolas Ancellin,‡ Sophie M. Bertrand,†,§ Ryan P. Bingham,† Paul S. Carter,† Chun-wa Chung,† Ian Churcher,† Nerina Dodic,‡ Charlène Fournier,† Peter L. Francis,† Andrew Hobbs,† Craig Jamieson,§ Stephen D. Pickett,† Sarah E. Smith,† Donald O’N. Somers,† Claus Spitzfaden,† Colin J. Suckling,§ and Robert J. Young†
†Medicines Research Centre, GlaxoSmithKline R&D, Gunnels Wood Road, Stevenage, Hertfordshire, SG1 2NY, U.K.
‡Les Ulis, Centre de Recherche, GlaxoSmithKline R&D, 25,27 Avenue du Québec, 91140 Villebon sur Yvette, France
§Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, U.K.
Abstract
The interfacial reduction of aqueous [PdCl4]2− at the interface with an organic solution of ferrocene has been characterised by X-ray absorption fine structure (XAFS) spectroscopy. Use of a liquid–liquid interface as a model for homogeneous nucleation permits control of the thermodynamic driving force for nucleation, through variation of the [PdCl4]2− and ferrocene concentrations in the bulk of the adjacent phases. We demonstrate that this approach permits characterisation of the system under conditions of (i) no particle nucleation, (ii) fast spontaneous nucleation of stable nanoparticles and (iii) an intermediate state, in which formation of metastable Pd sub-critical nuclei takes place. Analysis of the XAFS spectra in the metastable state revealed a stochastically fluctuating equilibrium in which Pd nuclei are constantly formed and re-dissolved, as evident from oxidation state fluctuations detected by the Pd XAFS. Supersaturation was evidently sufficient to induce nanoparticle formation but insufficient for nuclei to grow beyond the critical cluster size. We were able to maintain a system in this metastable state for several hours. Such sub-critical clusters are predicted by classical nucleation theory, but have not been detected except in liquid-cell TEM imaging and scanning electrochemical microscopy studies.
CrystEngComm, 2016, 18, 674 DOI: 10.1039/c5ce01883h
S.-Y. Chang,a Y. Gründer,‡ab S. G. Booth,b L. B. Molleta,a A. Uehara,§b J. F. W. Mosselmans,c G. Cibin,c V.-T. Pham,de L. Nataf,d R. A. W. Dryfeb and S. L. M. Schroeder¶*abc
a School of Chemical Engineering Analytical Science, University of Manchester, Manchester M13 9PL, UK. E-mail: [email protected]
b School of Chemistry, University of Manchester, Manchester M13 9PL, UK
c Diamond Light Source Ltd., Diamond House, Harwell Science Innovation Campus, Fermi Ave, Didcot, Oxfordshire OX11 0DE, UK
d Synchrotron SOLEIL, L'Orme des Merisiers, Saint-Aubin, BP48, 91192, Gif-sur-Yvette, France
e Center for Quantum Electronics, Institute of Physics, Vietnam Academy of Science and Technology, P.O. Box 429, Boho, 10000 Hanoi, Vietnam
† Electronic supplementary information (ESI) available: Open circuit potential, dispersive XAFS results and the MATLAB script for edge-step height fitting. See DOI: 10.1039/c5ce01883h
‡ Present address: Oliver Lodge Laboratory, Department of Physics, University of Liverpool, Liverpool L69 7ZE.
§ Present address: Division of Nuclear Engineering Science, Research Reactor Institute, Kyoto University, Asashironishi, Kumatori, Osaka, 590-0494, Japan.
¶ Present address: School of Chemical Process Engineering, Faculty of Engineering, University of Leeds, Leeds LS2 9JT.
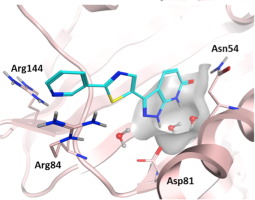
The lipid chaperone aP2/FABP4 has been implicated in the pathology of many immunometabolic diseases, including diabetes in humans, but aP2 has not yet been targeted for therapeutic applications. aP2 is not only an intracellular protein but also an active adipokine that contributes to hyperglycemia by promoting hepatic gluconeogenesis and interfering with peripheral insulin action. Serum aP2 levels are markedly elevated in mouse and human obesity and strongly correlate with metabolic complications. These observations raise the possibility of a new strategy to treat metabolic disease by targeting serum aP2 with a monoclonal antibody (mAb) to aP2. We evaluated mAbs to aP2 and identified one, CA33, that lowered fasting blood glucose, improved systemic glucose metabolism, increased systemic insulin sensitivity, and reduced fat mass and liver steatosis in obese mouse models. We examined the structure of the aP2-CA33 complex and resolved the target epitope by crystallographic studies in comparison to another mAb that lacked efficacy in vivo. In hyperinsulinemic-euglycemic clamp studies, we found that the antidiabetic effect of CA33 was predominantly linked to the regulation of hepatic glucose output and peripheral glucose utilization. The antibody had no effect in aP2-deficient mice, demonstrating its target specificity. We conclude that an aP2 mAb–mediated therapeutic constitutes a feasible approach for the treatment of diabetes.
Science Translational Medicine 23 Dec 2015: Vol. 7, Issue 319, pp. 319ra205 DOI: 10.1126/scitranslmed.aac6336
M. Furkan Burak1,*, Karen E. Inouye1, Ariel White1, Alexandra Lee1, Gurol Tuncman1, Ediz S. Calay1, Motohiro Sekiya1, Amir Tirosh1, Kosei Eguchi1, Gabriel Birrane2, Daniel Lightwood3, Louise Howells3, Geofrey Odede3, Hanna Hailu3, Shauna West3, Rachel Garlish3, Helen Neale3, Carl Doyle3, Adrian Moore3 and Gökhan S. Hotamisligil1,4,†
1Department of Genetics and Complex Diseases and Sabri Ülker Center, Harvard T.H. Chan School of Public Health, Boston, MA 02115, USA.
2Beth Israel Deaconess Medical Center, Boston, MA 02215, USA.
3UCB (Union Chimique Belge), 208 Bath Road, Slough, Berkshire SL1 3WE, UK.
4Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA.
↵†Corresponding author. E-mail: [email protected]
↵* Present address: Mount Auburn Hospital, Cambridge, MA 02138, USA. Science Translational Medicine 23 Dec 2015:
Week Commencing 18th April 2016
Abstract
Inhibitors of the ATPase function of bacterial DNA gyrase, located in the GyrB subunit and its related ParE subunit in topoisomerase IV, have demonstrated antibacterial activity. In this study we describe an NMR fragment-based screening effort targeting Staphylococcus aureus GyrB that identified several attractive and novel starting points with good ligand efficiency. Fragment hits were further characterized using NMR binding studies against full-length S. aureus GyrB and Escherichia coli ParE. X-ray co-crystal structures of select fragment hits confirmed binding and suggested a path for medicinal chemistry optimization. The identification, characterization, and elaboration of one of these fragment series to a 0.265 μM inhibitor is described herein.
Bioorganic & Medicinal Chemistry Letters Volume 26, Issue 4, 15 February 2016, Pages 1314–1318 doi:10.1016/j.bmcl.2016.01.009
Michael F. Mesleha, Jason B. Crossa, Jing Zhanga, Jan Kahmannb, Ole A. Andersenc, John Barkerc, Robert K. Chengc, Brunella Felicettic, Michael Woodc, Andrea T. Hadfieldc, Christoph Scheichb, Terence I. Moya, Qingyi Yanga, Joseph Shotwella, Kien Nguyena, Blaise Lippaa, Roland Dollea, M. Dominic Ryana
a Cubist Pharmaceuticals, 65 Hayden Ave., Lexington, MA 02421, United States
b Evotec AG, Manfred Eigen Campus, Essener Bogen 7, 22419 Hamburg, Germany
c Evotec (UK) Ltd., 114 Innovation Drive, Milton Park, Abingdon OX14 4RZ, UK
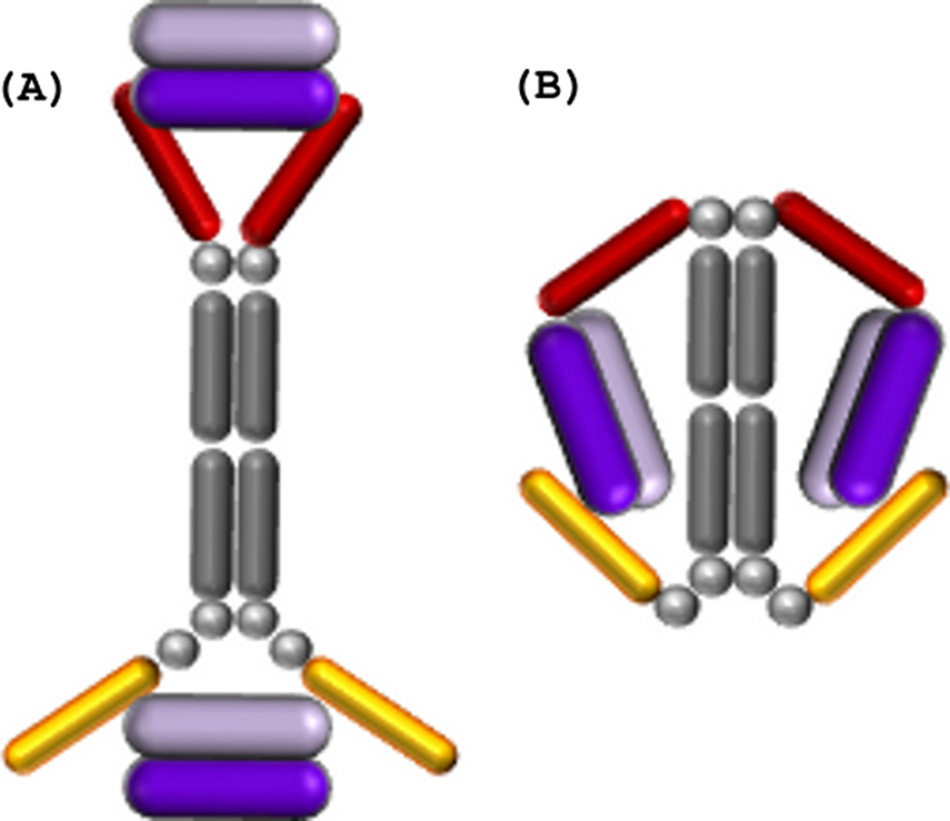
Abstract
A potent VEGF inhibitor with novel antibody architecture and antigen binding mode has been developed. The molecule, hereafter referred to as VEGF dual dAb (domain antibody), was evaluated in vitro for binding to VEGF and for potency in VEGFdriven models and compared with other anti-VEGF biologics that have been used in ocular anti-angiogenic therapeutic regimes. VEGF dual dAb is more potent than bevacizumab and ranibizumab for VEGF binding, inhibition of VEGF receptor binding assays (RBAs), and VEGF-driven in vitro models of angiogenesis and displays comparable inhibition to aflibercept (Eylea). VEGF dual dAb is dimeric, and each monomer contains two distinct anti-VEGF domain antibodies attached via linkers to a human IgG1 Fc domain. Mechanistically, the enhanced in vitro potency of VEGF dual dAb, in comparison to other antiVEGF biologics, can be explained by increased binding stoichiometry. A consistent model of the target engagement has been built based on the x-ray complexes of each of the two isolated domain antibodies with the VEGF antigen.
The Journal of Biological Chemistry, March 11 2016, 291, pages 5500-5511 doi: 10.1074/jbc.M115.691162
Adam Walker‡ , Chun-Wa Chung§ , Margarete Neu§ , Manish Burman‡ , Thil Batuwangala‡ , Gavin Jones‡ , Chi-Man Tang‡1, Michael Steward‡ , Michael Mullin¶ , Nadia Tournier¶ , Alan Lewis¶ , Justyna Korczynska‡¶, Vicky Chung , and Ian Catchpole‡2
‡ BioPharm Innovation,
§ Molecular Discovery Research,
¶ BioPharm Discovery, and BioPharm Process Research, GSK Medicine’s Research Centre, Stevenage, Herts SG1 2NY, United Kingdom

Abstract
C1ORF123 is a human hypothetical protein found in open reading frame 123 of chromosome 1. The protein belongs to the DUF866 protein family comprising eukaryote-conserved proteins with unknown function. Recent proteomic and bioinformatic analyses identified the presence of C1ORF123 in brain, frontal cortex and synapses, as well as its involvement in endocrine function and polycystic ovary syndrome (PCOS), indicating the importance of its biological role. In order to provide a better understanding of the biological function of the human C1ORF123 protein, the characterization and analysis of recombinant C1ORF123 (rC1ORF123), including overexpression and purification, verification by mass spectrometry and a Western blot using anti-C1ORF123 antibodies, crystallization and X-ray diffraction analysis of the protein crystals, are reported here. The rC1ORF123 protein was crystallized by the hanging-drop vapor-diffusion method with a reservoir solution comprised of 20% PEG 3350, 0.2 M magnesium chloride hexahydrate, 0.1 M sodium citrate pH 6.5. The crystals diffracted to 1.9 Å resolution and belonged to an orthorhombic space group with unit-cell parameters a = 59.32, b = 65.35, c = 95.05 Å. The calculated Matthews coefficient (VM) value of 2.27 Å3 Da-1 suggests that there are two molecules per asymmetric unit, with an estimated solvent content of 45.7%.
Acta Cryst. (2016). F72, 207-213 doi:10.1107/S2053230X16002016
Siti Nurulnabila A. Rahaman,a Jastina Mat Yusop,a Zeti-Azura Mohamed-Hussein,a,b Kok Lian Ho,c Aik-Hong Teh,d Jitka Watermane and Chyan Leong Nga*
aInstitute of Systems Biology, Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor, Malaysia,
bSchool of Biosciences and Biotechnology, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor, Malaysia,
cDepartment of Pathology, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia,
dCentre for Chemical Biology, Universiti Sains Malaysia, 11900 Bayan Lepas, Penang, Malaysia, and
eDiamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0DE, England

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Copyright © 2022 Diamond Light Source
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.

 Industrial Liaison Office
Industrial Liaison Office