
___________________________________
Industrial Liaison Group:
Tel: +44 (0) 1235 778797
E-mail: [email protected]
Following the successful implementation of Diamond’s fast_dp data processing pipeline, it became apparent that the bottleneck in structure analysis had been shifted from the data processing to the difference map calculation. While the data processing may be performed ab initio, the difference map calculation requires selection of an appropriate reference structure and preparation of the data. These interactive steps are followed by restrained refinement to generate an electron density map, which may be further inspected to determine the presence or absence of ligands. This process is straightforward for a few data sets but becomes challenging and time limiting in a high throughput environment. Rapid feedback about ligand binding is valuable for guiding subsequent data collection, especially when many samples of a particular protein-ligand complex are available.
In response to this situation, a new pipeline, DIMPLE, has been developed to:
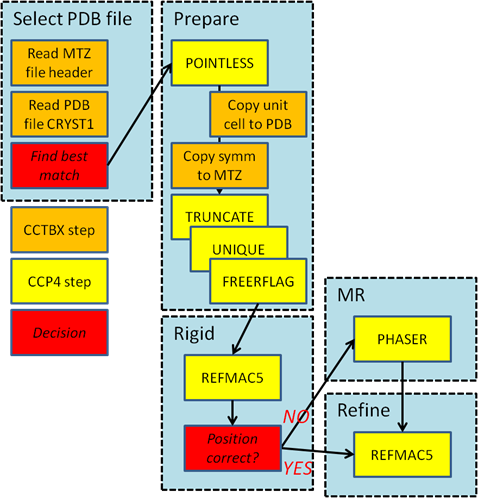
Figure 1: The workflow of Dimple.
DIMPLE’s workflow (Figure 1) is split into two phases: the selection of the reference model and the difference map calculation. The selection of the reference model compares the available models provided by the user to the output of fast_dp. The matching is based on the naming of the data, the pointgroup symmetry derived from the CRYST1 record in the coordinate file and the unit cell constants. Should the pointgroups from the model and from fast_dp be inconsistent the corresponding coordinate file will be ignored. If several coordinate files remain possible, the unit cell constants are compared and the closest matching file is selected. For orthorhombic lattices, permutations of a, b and c are allowed.
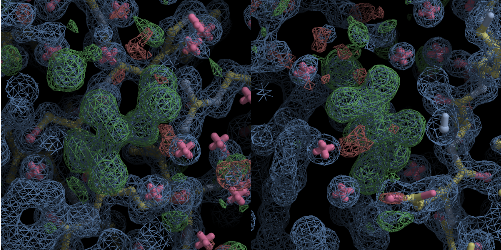
Figure 2: Two views of a tartrate ion identified automatically through DIMPLE analysis of data from a thaumatin sample.
For details on the Dimple pipeline, please contact [email protected] or [email protected] For assistance with the practicalities during data collection please first contact your local contact, who should be able to assist you.
How to name your model(s) and data:
To be considered as a candidate the name of the reference structure before the first full stop must appear in either the file name template or directory used for the data collection. For example abc.pdb will match /dls/…/in1234-5/abc/xyz_1_0001.cbf or /dls/…/in1234-5/xyz/abc_1_0001.cbf.
Where to place the model(s):
All available models, ideally with ligands and waters removed, have to be placed in /dls/…/in1234-5/processing/pdb
Multiple space groups option:
In situations where multiple spacegroups are possible, the suggestion is to have native files corresponding to each of the likely spacegroups and to name these files name.spacegroup.pdb (e.g. abc.P212121.pdb).
Where to find the results:
The results are available in your processed directory in either /dls/…/in1234-5/processed/xyz/fast_dp/dimple or /dls/…/in1234-5/processed/results/

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Copyright © 2022 Diamond Light Source
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.

 Industrial Liaison Office
Industrial Liaison Office