
Glucagon is a key hormone made by the pancreas, which raises blood sugar levels by acting on cell surface receptors. The interaction between glucagon and the glucagon receptor (GCGR) is therefore an important area of research for the treatment of diabetes.
The inhibition of glucagon signalling has been proposed as a method to reduce glucose production in the liver, but there is little understanding of the mechanism of action of recently developed small molecules that have been designed to target the GCGR. The aim of this study was to solve the crystal structure of one such inhibitor bound to the GCGR.
The X-ray structure of the GCGR bound with an inhibitor known as MK-0893 was determined using the Macromolecular Crystallography (MX) beamline, I04, at Diamond Light Source. The inhibitor was found bound to a previously unidentified site beyond a seven transmembrane helical bundle and extending into the lipid membrane of the cell. The unique binding site was confirmed by mutating key residues that were identified by the X-ray structure. This important new structural information could provide an opportunity for structure-based drug design for this class of receptors and might lead to a future series of novel inhibitors.
G protein-coupled receptors (GPCRs) are proteins which exist on the cell surface and contain a transmembrane domain (TMD) that undergoes conformational changes to couple extracellular signals (such as light, neurotransmitters, metabolites and hormones) to intracellular responses. GPCRs regulate many processes within cells, and hence are associated with many diseases. The class B secretin-like subfamily of GPCRs are validated drug targets for numerous diseases including osteoporosis, type 2 diabetes, depression, and anxiety.
Despite the increasing number of structures solved for transmembrane domains of class A GPCRs, relatively few are available for class B receptors. To date this was limited to the corticotropin-releasing factor receptor 1 (CRF1R) bound to the non-peptide antagonist CP-3763951 and the glucagon receptor (GCGR) in which the binding site of the small molecule antagonist NNC0640 could not be modelled2. Thus our understanding of the molecular basis of action of small molecules specifically targeting the TMD of class B receptors is limited.
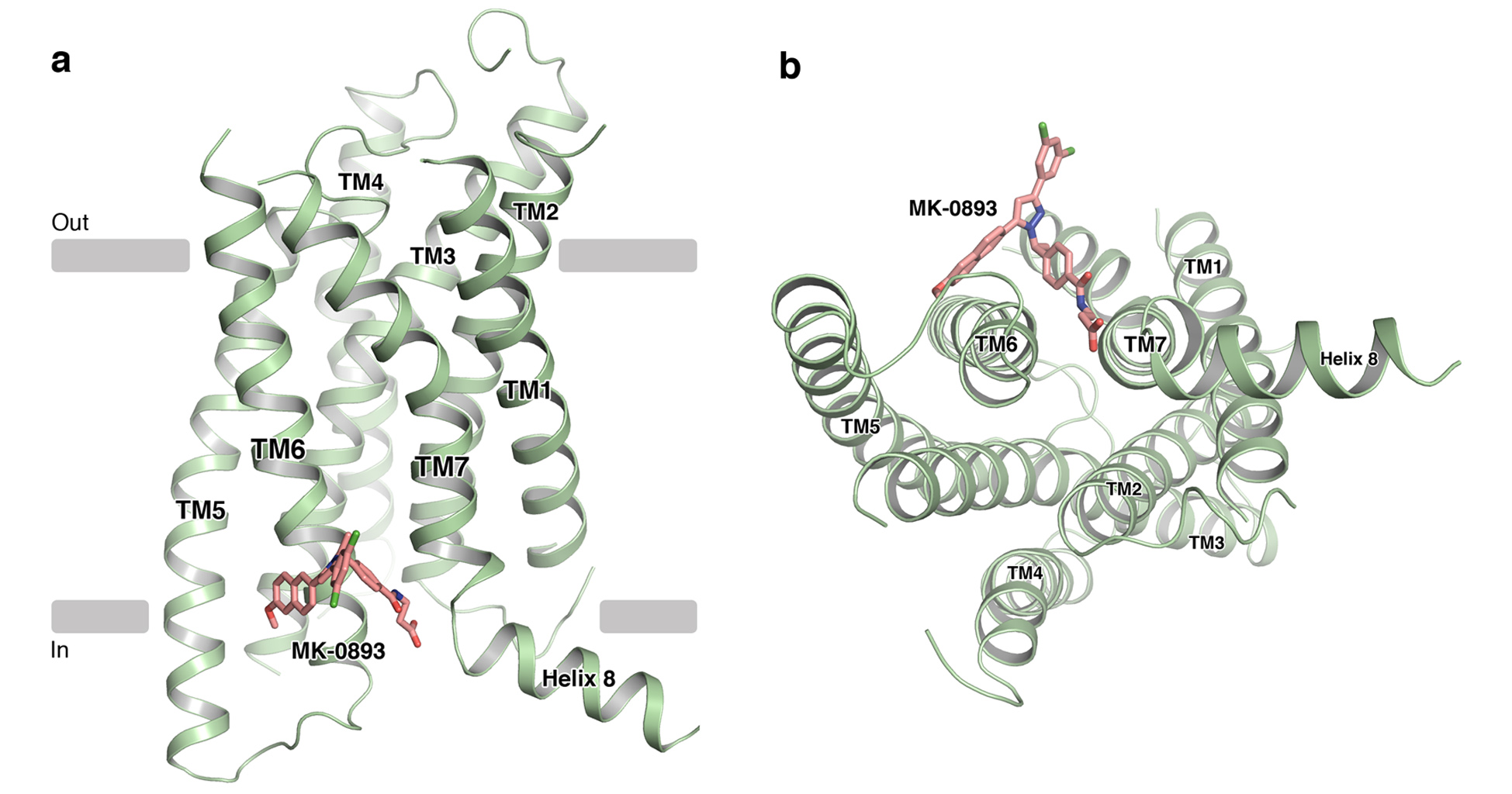
Figure 1: Structure of GCGR in cartoon representation as viewed (a) parallel to the membrane and (b) from the intracellular side. The antagonist MK-0893 is shown in stick representation with carbon atoms coloured pink, nitrogen atoms coloured blue, fluorine atoms coloured green and oxygen atoms coloured red.
Inhibition of signalling by the peptide hormone glucagon through the class B glucagon receptor has been demonstrated to improve glucose homeostasis in diabetic patients, and GCGR has been the subject of intensive studies to identify therapies for this increasingly prevalent condition. Over recent years several small molecule GCGR antagonists have entered clinical trials, however the binding site of these compounds on GCGR and the mechanism of antagonism of each were completely unknown. To address these questions the structure of human GCGR in complex with the small molecule antagonist MK-0893 using X-ray crystallography was determined.
The flexibility of GPCRs is essential for their function but problematic in terms of both their stability when extracted from the membrane environment, and their conformational heterogeneity, both of which can drastically hamper crystallisation. To mitigate this, 11 point mutations were introduced into GCGR, generating a stabilised receptor (StaR), increasing its thermal stability and concomitantly maintaining the receptor in an inactive conformation. The extra-cellular domain (ECD) was truncated and T4 lysozyme inserted into intracellular loop 2 (ICL2) to facilitate crystallogenesis. Small 40 μm oval shaped crystals of GCGR were obtained in lipidic cubic phase (LCP) and single crystals were cryo-cooled in liquid nitrogen and mounted for data collection. High quality diffraction data were collected on a Pilatus 6M detector using a 20 μm beam size on the I04 beamline at Diamond Light Source. Due to the intensity of the synchrotron X-ray beam and subsequent radiation damage, nine crystals were required to compile a complete dataset to 2.5 Å resolution, representing a substantial improvement on the previously published GCGR structure. The structure was solved using molecular replacement with independent search models consisting of the TMD of GCGR and T4 lysozyme from CRF1R (PDB ID: 4L6R and 4K5Y respectively).
Similar to previously determined GPCR structures, GCGR displays a canonical 7-transmembrane α-helical architecture (Fig. 1). The TMD of GCGR exhibits a very similar overall topology to the other published structure of GCGR with differences lying in the extracellular loop regions and a significant bulge in TM4 modulating inter-helical contacts and strengthening the TM4–TM3 interaction. As with the other class B receptor structure, human CRF1R, which was also solved in the inactive state, there are substantial differences between the orientation of the transmembrane helices of GCGR and those of class A receptors. Most notably, rotation of the extracellular portions of TM6 and TM7 leads to a more open configuration on the extracellular side that may be required to accommodate binding of large peptide hormones in class B GPCRs.
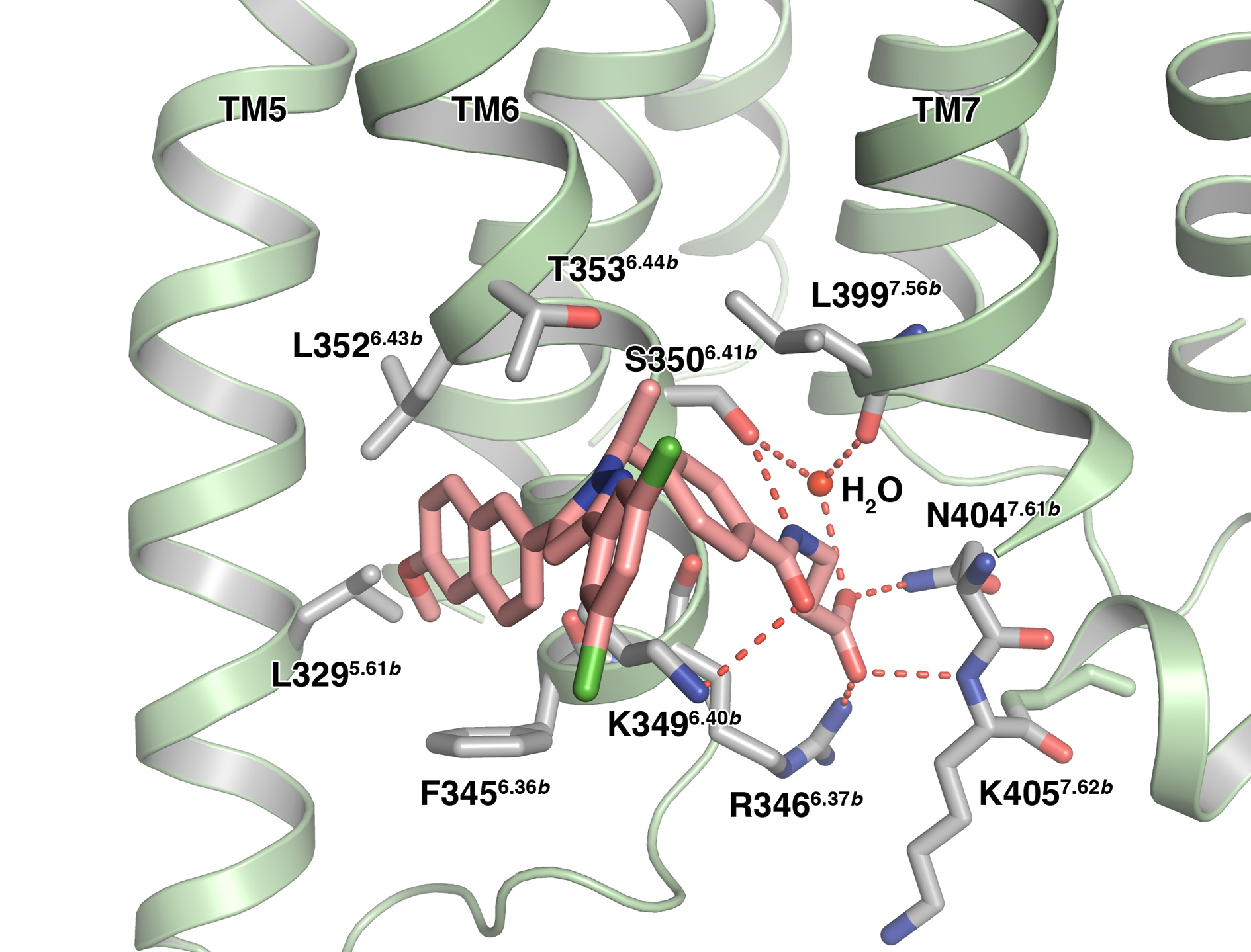
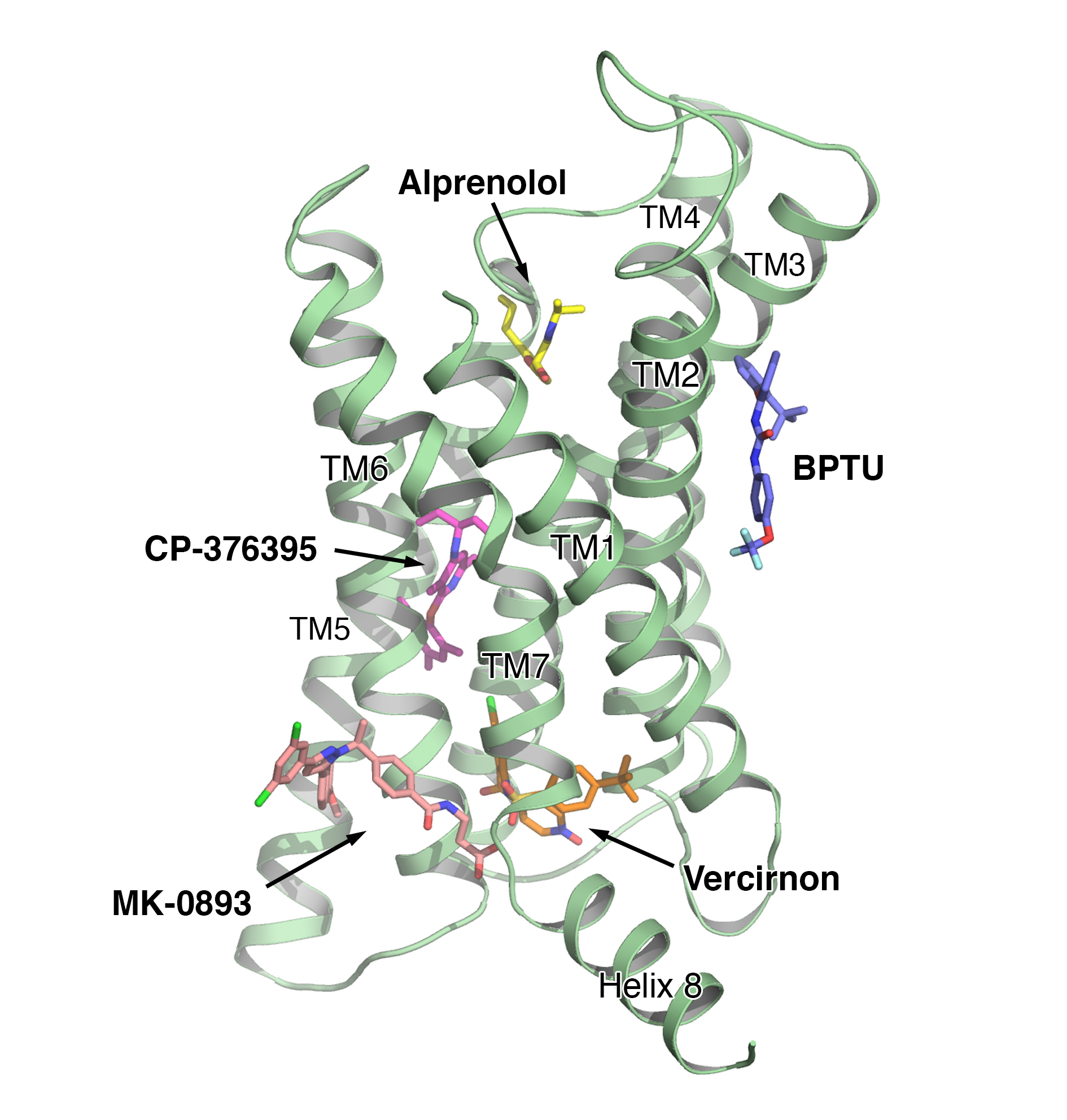
The most striking observation from this GCGR structure is that the small-molecule antagonist MK-0893 was found bound to an extra-helical site, close to the intracellular side of the receptor, but within the lipid bilayer (Fig. 1). The molecule could be clearly modelled bestriding TM6 and binding to two distinct regions: a hydrophobic surface on TM5, and a polar cleft adjacent to TM7, correlating with the physicochemical properties of the ligand (Fig. 2). These binding pockets have not been observed in any other GPCR structure solved and are distinct from the orthosteric region within the helical bundle where small-molecule ligands have been located in class A GPCRs. These class A binding sites largely overlap, but display variability in the depth of penetration into the transmembrane pocket. The GCGR site is also distinct from the antagonist binding site located in CRF1R, which lies deep within the helical bundle. An alternative site of extra-helical allosteric antagonism is observed in the class A receptor P2Y1 where the binding site for BPTU is located between TM1, 2 and 3 and likely inhibits the movement of TM3 that is critical in receptor activation3.
The MK-0893 binding mode suggests that the ligand acts as a clamp preventing the outwards rotation of TM6 that occurs during transition from the inactive to the active conformation required for G-protein coupling and subsequent signalling. Such movement has been observed in structures of class A GPCRs in the activated conformation, such as the ß2 adrenergic receptor bound to the Gs heterotrimeric G protein4.
So far, no active structures of class B receptors have been reported; however, the ability of MK-0893 to block signalling of GCGR points to a similar role for TM6 in class B receptor activation. The identification of an allosteric binding site in GCGR located outside the helical bundle adds to the diversity of interactions known to occur between GPCRs and ligands to influence receptor activation states (Fig. 3).
The structure of GCGR with MK-0893 provides further insight into the activation of class B receptors and offers an alternative mechanism by which receptor activity can be modulated. Strong sequence conservation of this binding site across human class B receptors indicates that the structural information generated could provide opportunities in small molecule drug design and discovery across members of this medically important family of GPCRs.
References:
Funding acknowledgement:
Funded solely by Heptares Therapeutics Ltd.
Corresponding author:
Dr Stacey Mary Southall, Heptares Therapeutics Ltd., [email protected]
Related publication:
Jazayeri A, Doré A, Lamb D, Krishnamurthy H, Southall S, Baig A, Bortolato A, Koglin M, Robertson N, Errey J, Andrews S, Teobald I, Brown A, Cooke R, Weir M, Marshall, F. Extra-helical binding site of a glucagon receptor antagonist. Nature 533, 274-277, doi: 101038/nature17414 (2016).
Publication keywords:
X-ray crystallography; G protein-coupled receptors

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Copyright © 2022 Diamond Light Source
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.

 Science
Science