
Broad-spectrum antibiotics are designed to combat a wide range of bacteria; however, the same therapeutic approach cannot be applied to viruses. Currently, viral infections need to be treated individually with specific antiviral drugs, but a novel therapeutic strategy has been developed to inhibit a shared viral enzyme known as α-glucosidase II. This enzyme is responsible for folding viral surface glycoproteins to ensure the viral envelope is properly constructed during replication.
To further understand this potential broad-spectrum antiviral target, its three-dimensional structure was solved using the Macromolecular Crystallography beamlines (I02, I03, I04 and I04-I) at Diamond Light Source. The enzyme was characterised both alone and in combination with ligands and antiviral iminosugars (including two that are undergoing clinical trial for dengue fever).
The first ever crystal structure of a large fragment of α-glucosidase II yielded a number of significant observations: the quaternary structure of the enzyme, the stages of the catalytic cycle, and the binding mechanism of the antiviral iminosugars. Furthermore, the structural insights from the study suggested that the potency and selectivity of potential antivirals could be enhanced by targeting a conserved ring of aromatic residues between the α-glucosidase II +1 and +2 subsites. This knowledge could provide a template for designing highly effective broad-spectrum antivirals.
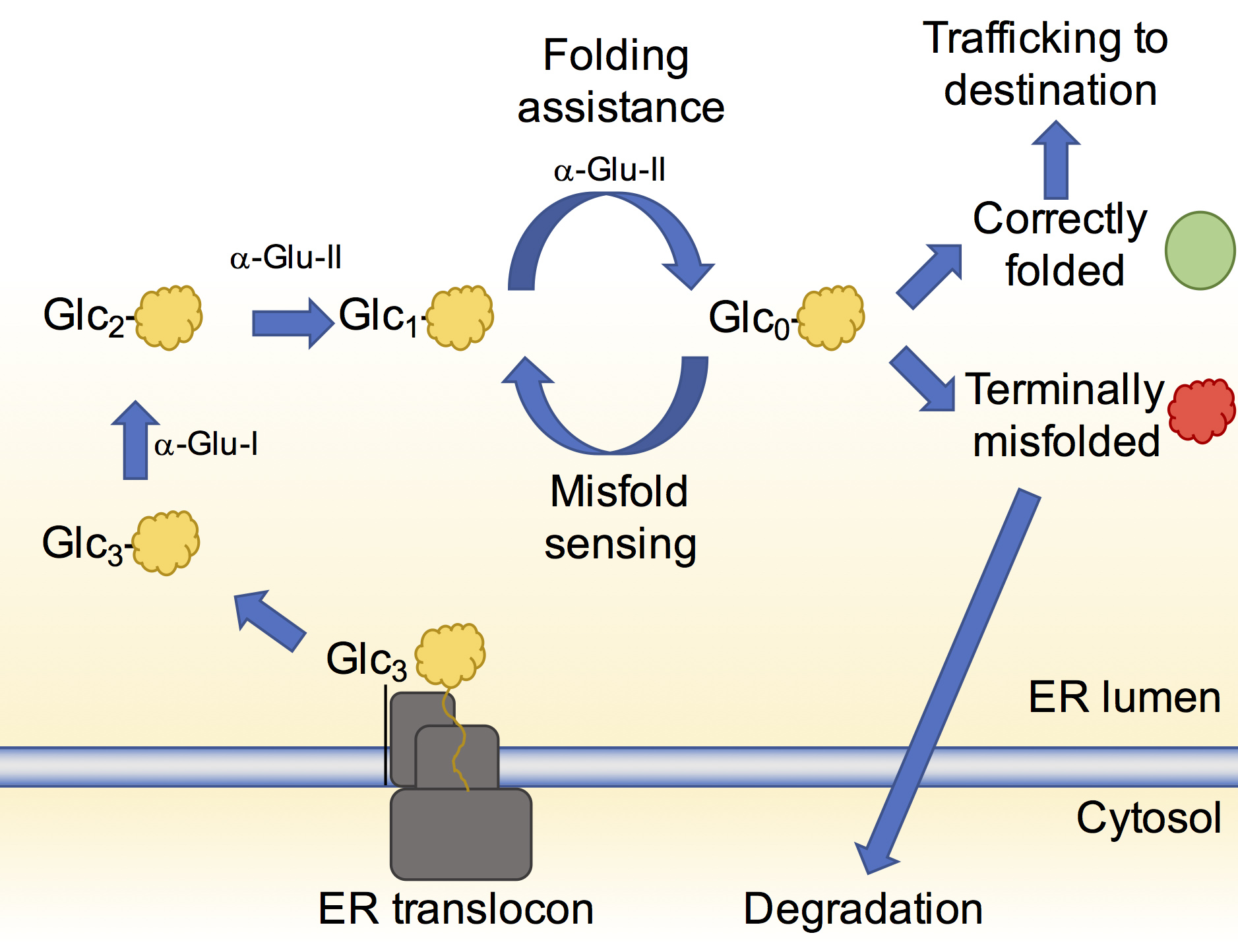
The proteins encoded by the genome in eukaryotes usually undergo chemical modifications during or after their production. The most common of these modifications is N-linked glycosylation, which attaches a complex series of sugars to asparagine residues in the endoplasm reticulum (ER) lumen resulting in what is called a glycoprotein.
It is estimated that at least 20% of genes code for proteins that acquire N-linked glycans1. The roles of these glycans vary but at the very initial stages of the glycoprotein being formed they are crucial for helping the glycoprotein attain its native three-dimensional structure. The glycans once modified by specific enzymes, act as flags to indicate the folding state of the glycoprotein. A flagged glycan on a glycoprotein enables its association with chaperones and other enzymes that in turn aid its folding and correct mistakes that may have occurred (Fig. 1). This glycoprotein folding control pathway is called the calnexin cycle2.
When the glycoprotein has achieved its native state, removal of the flag from the glycan allows it to move onto its correct destination in, or out, of the cell. If there is a terminal defect in the glycoprotein, the glycan reflects this and the terminally misfolded glycoprotein is redirected to the cytosol for degradation.
The most important flag that an N-linked glycan can carry at this stage is a glucose moiety. Soon after N-linked glycans are attached to a glycoprotein, two ER-resident enzymes, ER α-glucosidase I and II, sequentially remove two of the three glucose residues on the glycan3. When a glycoprotein is left with only a single glucose on its glycan, it can interact with the chaperones helping it to fold. While the structure of ER α-glucosidase I from yeast was determined in 2013, ER α-glucosidase II (α-Glu-II) had eluded structural characterisation for some time. It was thought to closely resemble enzymes in the intestinal tract that break down sugars and enable glucose absorption. The full-length enzyme exists as a heterodimer consisting of a catalytic a-subunit and a regulatory β-subunit (Fig. 2a) that is resistant to crystallisation. However, mixing mouse α-Glu-II with a small amount of a protease, allowed us to purify and crystallise a slightly smaller and stable α-Glu-II fragment. Our crystal structure confirmed that the catalytic subunit was similar to ones of intestinal glucosidases (Fig. 2b). However, a protease resistant domain of the regulatory subunit remained attached, revealing two low-density lipoprotein receptor type A (LDLRa) domains and the site of dimerisation of the enzyme (Fig. 2b in blue).
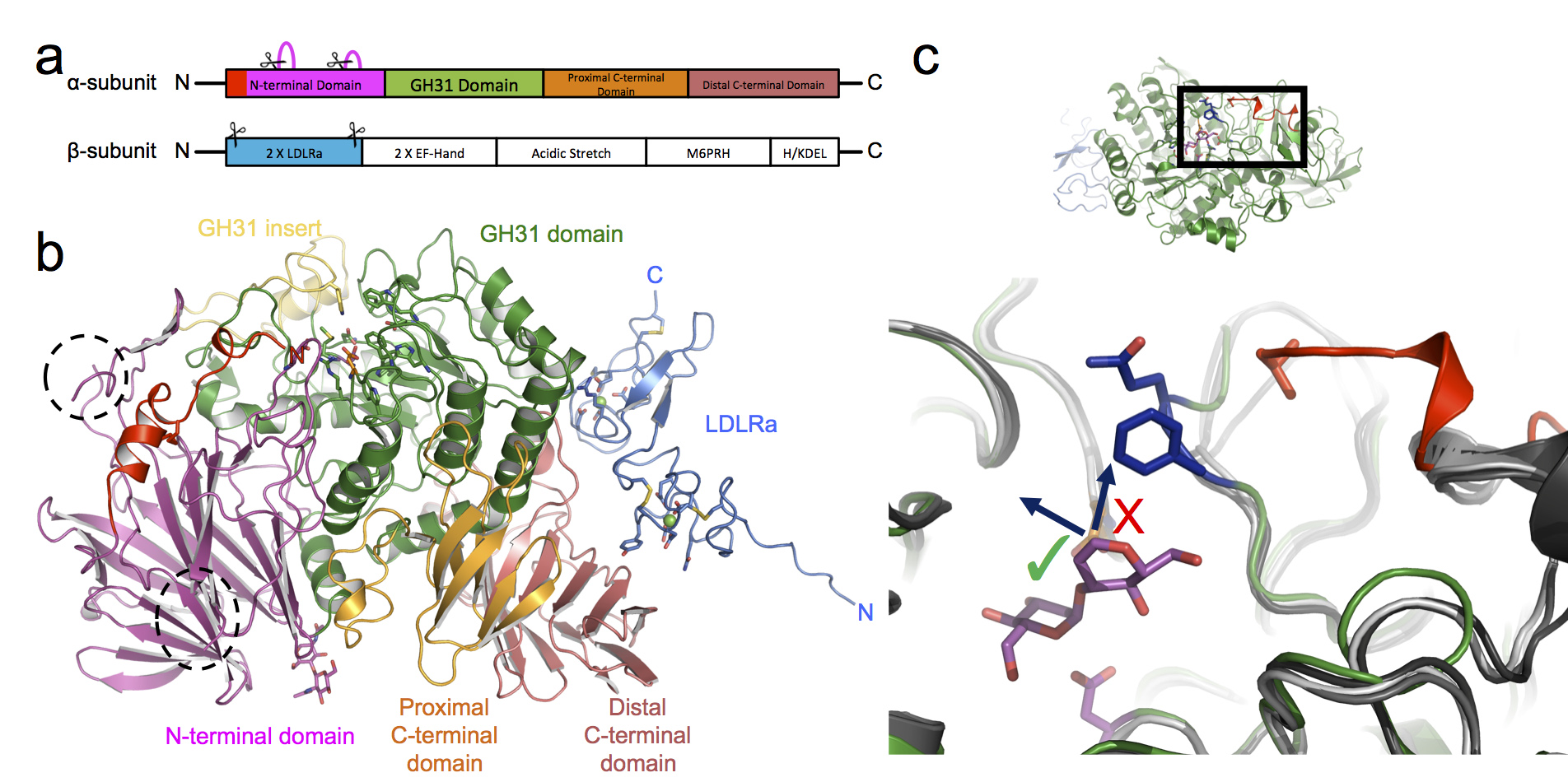
Figure 2: Structure of the proteolysed mouse α-Glu-II. (a) Linear domain arrangement of the α-subunit and β-subunit. Scissors indicate the parts of the enzyme removed by proteases. (b) The 3D architecture of the heterodimeric enzyme coloured by domain. (c) A top-down view into the active site with the bound pseudo-substrate (in magenta). The α-Glu-II-specific selectivity loop (blue residues) protrudes into a volume which in other related glycosidases (shades of grey) do not have this insertion. The result is that longer glycans can only extend in a particular direction (arrow with green tick).
By soaking in a number of different carbohydrates into the crystals, and collecting X-ray data on four of the Macromolecular Crystallography beamlines at Diamond: I02, I03, I04, and I04-1, a series of structures of the mouse α-Glu-II fragment in complex with these ligands was determined. These snapshots of the catalytic cycle illustrate the mechanism of α-Glu-II and allow a comparison between this enzyme and the intestinal glucosidases. The core of the catalytic site is extremely similar but further to the rim there are some striking differences that indicate where the basis of substrate specificity arises. A loop, which we termed the ‘selectivity loop’, excludes polysaccharide ligands from extending in the same direction as those found in the intestinal glucosidases (Fig. 2c).
Many enveloped viruses, including influenza, hepatitis C virus, HIV and dengue virus among others, contain N-linked glycoproteins on the surface of the viral particles. During infection, these glycoproteins are subject to the same folding pathway that the host-cell glycoproteins undergo. Disrupting entry into the calnexin cycle by inhibiting either of the ER a-glucosidases can reduce the infectivity or assembly of new viral particles4. This is an effective antiviral strategy for a number of viruses in vitro and in vivo.
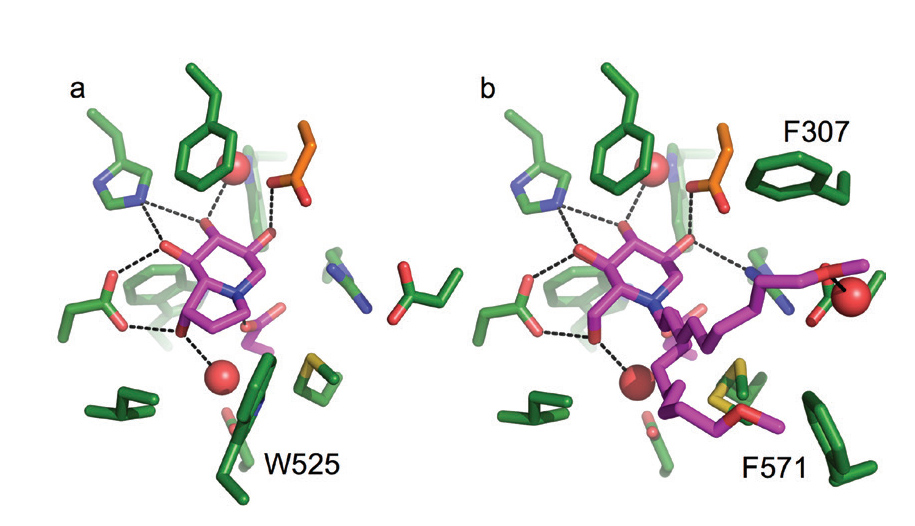
Figure 3: Crystal structures of the active site of the murine α-Glu-II fragment bound to clinically relevant inhibitors castanospermine (a) and MON-DNJ (b). α-Glu-II residues represented in green stick, inhibitors in magenta, ordered water molecules shown in red spheres, and hydrogen bonds represented with black dashed lines.
Two inhibitors (called castanospermine and MON-DNJ) are currently in clinical trials for the treatment of acute dengue fever. These inhibitors are part of a family of glucomimetics called iminosugars. The crystal structures, in this study, of castanospermine and MON-DNJ bound to the mouse α-Glu-II fragment (Fig.3 a-b) highlight their modes of binding. MON-DNJ (Fig 3b) has a long alkyl chain that improves its pharmacokinetics but in the crystal structure appears to adopt two conformations. The exclusion of a tryptophan (W525) that contributes to the active site for substrate binding may be important for the increase in potency of MON-DNJ over castanospermine. One of the conformations of the alkyl tail of MON-DNJ sits between a fork made by two phenylalanine residues (F307 and F571). Targeting these discovered features may allow development of newer more targeted inhibitors. Furthermore, these inhibitors are unfortunately also inhibitors of other glucosidases in the body, causing unwanted albeit minimal side effects. Our crystal structures now enable a rational drug discovery approach for more selective inhibitors. XChem fragment screening at DLS is ongoing, which might enable discovery of a completely new chemical series with greater levels of selective inhibition of α-Glu-II for more efficacious antiviral compounds.
References:
Funding acknowledgement:
A.T.C. was funded by a Wellcome Trust 4-Year Studentship (097300/Z/11/Z). Oxford Glycobiology Institute Endowment.
Corresponding authors:
Dr Alessandro Caputo, alessandro.caputo@bioch.ox.ac.uk, Dr Pietro Roversi, pietro.roversi@bioch.ox.ac.uk , Prof Nicole Zitzmann, University of Oxford, nicole.zitzmann@bioch.ox.ac.uk
Related publication:
Caputo AT, Alonzi DS, Marti L, Reca I-B, Kiappes JL, Struwe WB, Cross A, Basu S, Lowe ED, Darlot B, Santino A, Roversi P, Zitzmann N. Structures of mammalian ER α-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proceedings of National Academy of Sciences of the United States of America. 113(2), E4630-E4638, doi:10.1073/pnas.1604463113 (2016).
Publication keywords:
Broad-spectrum antiviral; ER α-glucosidase II; Eukaryotic secretion; Glycoprotein folding; Iminosugar

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Copyright © 2022 Diamond Light Source
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.

 Science
Science