All living cells require energy, usually provided in the form of ATP, to carry out fundamental processes like movement and growth. Therefore, cells have to balance energy supply with the demand. AMP-activated protein kinase (AMPK) has emerged as a central component of a signalling pathway involved in regulating intracellular energy homeostasis. When ADP and AMP levels increase, concomitant with a fall in ATP levels, AMPK is activated by phosphorylation on threonine-172 within the catalytic a subunit by upstream kinases including LKB1 and calcium/calmodulin protein kinase kinase (CaMKK) ß. Once activated, AMPK responds by phosphorylating downstream targets, the net effect of which is to ‘switch off’ ATP-consuming processes and ‘switch on’ ATP-producing processes. When the levels of nucleotides are restored to normal physiological concentrations, AMPK is inactivated by an unknown protein phosphatase(s). Major advances have been made over the last five years in understanding the molecular mechanism of how AMPK acts as an energy sensor. The ability to express the AMPK heterotrimer (catalytic a subunit and regulatory ß and ? subunits) in bacteria has provided a useful tool to study the regulation of AMPK in vitro. Part of this work has involved determining the X-ray crystal structure of AMPK using the Diamond Light Source and is the most complete structure to date of the enzyme.
Our previous studies demonstrated that a key physiological mechanism for activating AMPK is the ability of AMP to prevent dephosphorylation of threonine-172 and inactivation of AMPK.1 AMP can also allosterically activate AMPK, although this effect is modest at around 2-fold. A structure of the core complex of mammalian AMPK (C-terminal a, C-terminal ß and full length ?) demonstrated that AMP binds to the AMPK ? subunit between the cystathione-ß-synthase (CBS) motifs, of which there are four.2 Three molecules of AMP were shown to bind to AMPK, two of which can exchange with ATP, which leads to inhibition of the allosteric activation and protection from dephosphorylation by AMP. Thus, AMP binding to the ? subunit leads to activation of AMPK. Under optimal conditions, mammalian cells maintain ATP at a high level relative to ADP and AMP. The concentrations of free adenine nucleotides in mammalian cells lie in the range of 3000-8000 µM for ATP, 50-200 µM for ADP and 0.5-5 µM for AMP. This raised the question of how relatively low concentrations of AMP can compete with high concentrations of ATP to activate AMPK. We have extended our previous findings to demonstrate that ADP can also protect AMPK from dephosphorylation, although ADP does not allosterically activate the enzyme.3 Since the concentration of ADP is up to 400-fold higher than AMP, ADP will be more successful at competing with ATP than AMP. Therefore, the fact that ADP protects AMPK from dephosphorylation suggests that this is likely to represent an important physiological mechanism for regulating the activity of the enzyme.
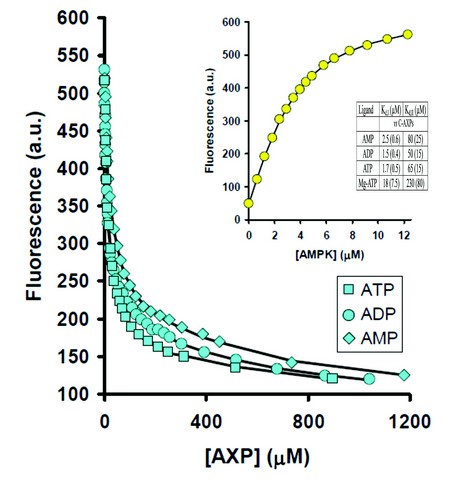
Figure 1: Displacement of coumarin-ATP by AXPs monitored using fluorescence at 470 nm. Inset: titration of coumarin-ATP with AMPK and dissociation constants (Kd) for the binding of AXPs. Values are reported as the mean (±s.d.) determined from at least three independent measurements.
We characterised the binding properties of the three nucleotides to full-length active AMPK using the coumarin adducts of ATP and ADP as fluorescent reporters of nucleotide binding. This allowed the binding parameters for the unlabeled nucleotides to be derived by performing competition experiments (Fig. 1). It was demonstrated that all three nucleotides bind with similar affinities to AMPK. Under physiological conditions, most of the ATP is coordinated to Mg2+, and the majority of AMP and ADP is not, therefore, we also measured nucleotide binding of Mg.ATP. Our data show that Mg.ATP binds up to 10-fold weaker than ATP and importantly, AMP and ADP. Taken together, this provides clues to how AMP and ADP can activate AMPK under physiological concentrations of Mg.ATP.
An interesting finding to emerge from the binding studies is that the two exchangeable nucleotide binding sites have different affinities. Binding at the tighter of the two sites is at least 30-fold stronger than at the weaker site. The concentrations of AMP required to achieve allosteric activation is consistent with binding at the tighter site, whereas the concentrations of AMP and ADP that protect AMPK from dephosphorylation are consistent with binding at the weaker site. We also carried out co-crystallization of the core complex with one molar equivalent of ADP. The resulting electron density map showed full occupancy of ADP at site-1 and no detectable density at site-3, identifying site-3 as the weaker binding site. Therefore, we can assign the allostery to AMP binding at the tighter site-1 and protection against dephosphorylation is mediated by AMP or ADP binding at the weaker site-3.
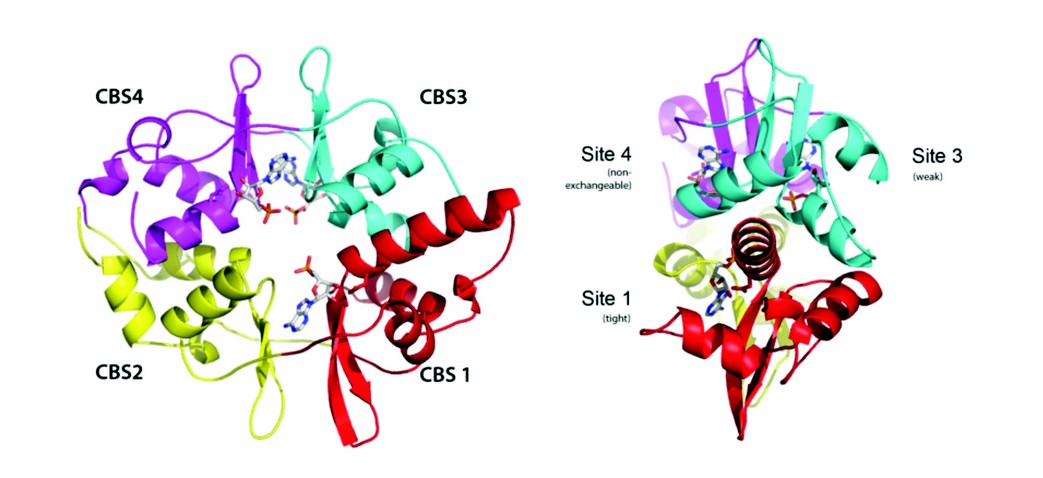
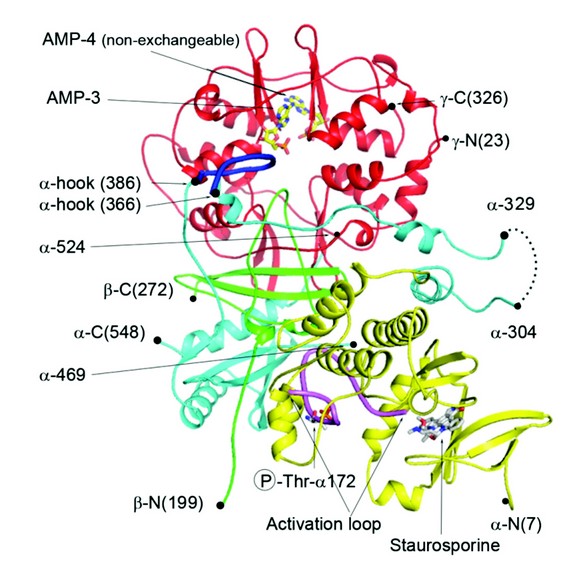
Figure 2: Structure of AMPK. A, Ribbon representation of AMPK ? subunit with three AMP molecules bound. The CBS domains and numbering of each site is also shown. B, Ribbon representation of the active complex highlighting the two AMP molecules bound, the phosphorylated Thr172 and the a hook (heavier lines, dark blue).
We have used X-ray crystallography to determine the structure of an active form of AMPK and is the most complete structure to date, only lacking the glycogen binding domain of the N-terminus of the regulatory ß subunit (Fig. 2). One of the steps towards diffraction quality crystals involved engineering in a pair of protease recognition sites at both ends of a flexible loop in the C-terminal domain of a. We had previously observed that this loop was disordered in our crystal structure of the regulatory fragment of AMPK and reasoned that being able to excise this loop, by specific protease treatment, might enhance crystallization as observed in other systems. It was also necessary to phosphorylate the AMPK complex with CaMKKß and incubate the enzyme with the kinase inhibitor staurosporine and AMP.
The structure revealed that significant contacts are made by the activation loop of the kinase domain predominantly with the C-terminus of the ß subunit. The fact that the activation loop mediates the interaction of the kinase domain with the regulatory fragment means that the phosphorylated Thr-172 is protected from access by phosphatases. Another component of the a subunit/regulatory interaction is provided by a part of the segment of the a chain which links the N-terminal kinase domain to the C-terminal regulatory fragment, which we have termed the a-hook. The a-hook interacts with the ? subunit at the exchangeable binding site-3 that we have assigned as the weaker of the two sites that is responsible for mediating AMP/ADP protection against dephosphorylation. Based on superposition of our earlier structures of the regulatory fragment of AMPK in complex with ADP and Mg.ATP and the structure presented here, we think that the a-hook sequence cannot interact with site-3 when Mg.ATP is bound mainly due to the change in conformation of ? arginine-69 which would generate a steric clash with the a-hook.
This structure reveals the mechanism for how AMP or ADP, but not ATP, protect Thr-172 from dephosphorylation. Binding of AMP or ADP allows the interaction of the a-hook which acts to restrict the flexibility of the preceding a linker region and in so doing promotes the interaction of the kinase domain and activation loop with the regulatory fragment. This interaction would therefore act to protect Thr-172 against dephosphorylation only when AMP and ADP are bound, but not when ATP is bound.
Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. Nature. 2011 Apr 14;472(7342):230-3
References
- Sanders, M.J., et al., Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J, 2007. 403(1): p.139-48.
- Xiao, B., et al., Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature, 2007. 449(7161): p. 496-500.
- Xiao, B., et al., Structure of mammalian AMPK and its regulation by ADP. Nature, 2011. 472(7342): p. 230-3.
Acknowledgements
Work in both laboratories is supported by the Medical Research Complex and we gratefully acknowledge Diamond Light Source for synchrotron access.



 Science
Science