
Chirality, the property of an object to exist as distinguishable mirror image forms (known as enantiomers), is ubiquitous. Intriguingly, the chemistry of life is intrinsically homochiral, as biological molecules such as sugars, amino acids and DNA exist almost exclusively as only one enantiomer. Identifying potential mechanisms for such asymmetry is therefore of tremendous interest, not least because those mechanisms may also allow more efficient asymmetric synthesis of pharmaceuticals. In a recent X-ray photoemission study of chiral gold nanoparticles, conducted on the Nanoscience beamline I06, we discovered a pronounced electron scattering mechanism that could provide an efficient new route to enantioselective chemistry.
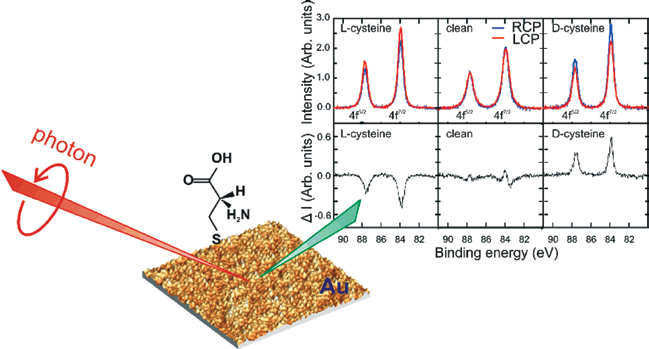 |
| Figure 1: Strong circular dichroism is observed in the Au 4f7/2, 4f5/2 core level photoemission spectra collected from cysteine-treated nanoparticulate gold. Left panels: Au reconstructed using L-Cys. Middle panels: clean Au. Right panels: Au reconstructed using D-Cys. Spectra were collected using right (blue, RCP) and left (red, LCP) circularly polarized X-rays at 300K. The difference spectra are shown in the lower panels, where a slight drift in energy, rather than dichroism, accounts for the features in the clean spectrum. Also shown is an atomic force microscopy image along with a schematic of the experimental geometry |
One of the earliest suggestions for natural asymmetric production of enantiomers was the initiation of photochemical reactions by circularly polarised light [1], but this is typically a weak effect. More recently, low-energy, spinpolarised ballistic electrons have also been found to scatter differentially from chiral materials, thereby illustrating the type of asymmetry required to promote the formation, or degradation, of one enantiomer over another [2][3]. Nevertheless, most electron-scattering experiments tend to show either a rather weak asymmetry in absolute intensity or an asymmetry only in the angular distribution of scattering. In contrast, we observed strong differential scattering of electrons carrying orbital angular momentum. Specifically, photoelectrons that are ejected from specific core-level electronic states with circularly polarised light are known to have a helical phase component that imbues chirality independent of any electronic spin polarization [4]. We found such photoelectrons to be filtered by an interface formed by etching a thin film of gold nanoparticles with the chiral molecule cysteine. The resultant asymmetry in scattering depends upon both the photon polarization and the surface chirality. Most importantly, because the effect was measured from a random distribution of supported gold nanoparticles, we were able to discount a simple angular redistribution of scattered electrons and concluded that the asymmetry was truly in the absolute, energy-resolved photoelectron yield. As such, the effect could be of broad applicability.
We created a chiral interface by adsorbing aqueous cysteine [Cys = HSCH2CH(NH2)CO2H] onto a thin (~20nm) sputter-deposited film of polycrystalline gold. Atomic force microscopy of the fresh Au film indicated an isotropic distribution of flattened nanoparticles with a peak-to-peak roughness below 5 nm (Fig. 1). Cys adsorption, which is known to cause substantial “etching” and redeposition of gold [5][6] [7], was then found to flatten and enlarge the nanoparticles in a process similar to sintering. On flat surfaces, Cys is known to form a mobile Cys-Au moiety that can diffuse and re-order within chiral overlayers, mediated by inter-moiety hydrogen bonding [5]. Similarly, Cys and related molecules have long been used to create chiral conformations of solvated gold nanoparticles [8][9] and to re-facet crystal surfaces [10]. Thus, the outer layers of our polycrystalline film are expected to adopt chiral adatom arrangements. We then used spectroscopic photoemission electron microscopy (PEEM) at the Nanoscience beamline (I06) to study x-ray photoemission of the gold core-level 4f7/2, 4f5/2 states. An advantage of this system is the ability to energy-disperse spectra onto a pixilated detector and thereby capture a complete spectrum in a single image without scanning the kinetic energy.
Fig. 1 summarises our results and illustrates a previously unobserved, but strong photoemission circular dichroism (CD) – a differential response to left- and right circularly polarised light. The data are x-ray photoemission spectra collected from Cys-exposed Au nanoparticles and recorded using an incident photon energy of 690 eV. The three upper panels present results for L-Cys, the clean surface and D-Cys, with the signal collected using left-circularly polarised light (LCP) in red and using right-circularly light (RCP) in blue. The data show a strong circular dichroism upon a reversal of X-ray polarization and upon changing Cys enantiomers, whilst none is observed from clean Au. Interestingly, there is no clear spin-dependence in the dichroism since the relative intensities of 4f7/2 and 4f5/2 peaks, which have opposite spin states [11], are constant within experimental precision. This spin-independence contrasts with other systems [12], especially those measured at lower electron energies [13]. It allows us to eliminate Cys-induced ferromagnetism in surface Au atoms as an explanation for our data, which has been observed previously, but at a very weak level [14]. The CD of Fig. 1 is unusual because it is: (i) large; (ii) derived from electrons which predominately originate from the achiral bulk of the Au; (iii) measured from an azimuthally isotropic polycrystalline film; and (iv) observed along the sample normal in an achiral geometry. As such, it is inconsistent with known dichroic mechanisms. For example, the dominance of bulk signal allows us to discount dichroic photoexcitation of chiral centres, which in any case is typical of molecular systems at substantially lower electron energies and is predicted to be weak [15,16]. In our case, the photoelectron mean free path implies that around 80% of the Au XPS originates from emitter atoms below the outermost atomic layer, where the local environment cannot be chirally co-ordinated. Alternatively, known mechanisms for angular shifts in photoelectron emission can also be discounted. Simple geometric arguments indicate that the polycrystalline nature of our sample will azimuthally-average any photoelectron diffracted intensity, such that any dichroic rotations would not be observed.
The chirality arising from the helical phase of electrons with orbital angular momentum is often overlooked and has not previously been shown to show dichroism. However, we propose a differential transmission probability for these intrinsically-chiral photoelectrons as they pass through the chirally-sculptured interface, leading to a chiral dependence of the measured core-level photoelectron yield. In our experiment, the helical phase arises from photoexcitation using circularly polarised light. The filtering effect must then arise from an asymmetric, inelastic scattering of the rotating photoelectronic wavefront from the chirally reconstructed surface, with intensity being lost to the secondary electron background. Such chiral filtering has similarities with magneto-chiral anisotropies, where the cyclotron trajectories of electrons moving within a magnetic field scatter from extended chiral structures including crystallographic screw dislocations [17]. Importantly, the large difference in electron energies between such magnetoresistance study and the present work suggests that this new effect operates over a wide electron energy range, an aspect that we now plan to probe in more detail and using a number of core-level states. A particularly exciting development is the recent demonstration of free electron beams carrying orbital angular momentum [18]. Given the unexpectedly strong scattering asymmetry observed in our study, we now look forward to assessing the impact of such electrons on chiral chemical synthesis.
References
[1] W.A. Bonner, J.M. Greenberg, E. Rubenstein, Origins of Life and Evolution of the Biosphere 29, 215 (1999).
[2] S. Mayer, J. Kessler, Phys. Rev. Lett. 74, 4803 (1995).
[3] C. Nolting, S. Mayer, and J. Kessler, J. Phys. B: At. Mol. Opt. Phys. 30, 5491 (1997).
[4] H. Daimon, T. Nakatani, S. Imada, and S. Suga, J. Electron Spectrosc. Relat. Phenom. 76, 55 (1995).
[5] M .Yu, N. Bovet, C.J. Slatterley, S. Bengió , et al., Phys Rev. Lett. 97, 166102 (2006).
[6] A.S. Dakkouri, D.M. Kolb, R. Edelstein-Shima, and D. Mandler, Langmuir, 12, 2849 (1996).
[7] R.R. Nazmutdinov, J. Zhang, T.T. Zinkicheva, et al., Langmuir, 22, 7556 (2006).
[8] P.D. Jadzinsky, G. Calero, C.J. Ackerson, et al., Science, 318, 430 (2007).
[9] C. Gautier and T. Bürgi, J. Am. Chem. Soc. 130, 7077 (2008).
[10] X .Zhao, J. Am. Chem. Soc. 122, 12584 (2000).
[11] J.G. Tobin, S.W. Yu, T. Komesu, et al., Europhys. Lett. 77,17004 (2007).
[12] R.A. Rosenberg, M. Abu Haija, and P.J .Ryan, Phys. Rev. Lett. 101, 178301 (2008).
[13] K. Ray, S.P. Ananthavel, D.H. Waldeck, and R. Naaman, Science, 283, 814 (1999).
[14] M. Schunack, E. Lægsgaard, I. Stensgaard, et al. Angew. Chem., Int. Ed. 4, 2623 (2001).
[15] J.W. Kim, M. Carbone, J.H. Dil, et al., Phys. Rev. Lett. 95, 107601 (2005).
[16] F. Allegretti, M. Polcik, D.I. Sayago, et al., New J. Phys. 7, 109 (2005).
[17] G.L.J.A. Rikken, J. Fölling, and P. Wyder, Phys. Rev. Lett., 87, 236602 (2001).
[18] M. Uchida and A. Tonomura, Nature, 464, 737 (2010).
Principal Publications and Authors
Asymmetric photoelectron transmission through chirallysculpted, polycrystalline gold, D.A. MacLaren, J. Johnston, D. A. Duncan, H. Marchetto, S. S. Dhesi, N. Gadegaard and M. Kadodwala, Phys. Chem. Chem. Phys. 11, 8413-8416, (2009).
Funding Acknowledgement
The Scottish Universities Physics Alliance. Some text and figures reproduced by permission of the PCCP Owner Societies.

Diamond Light Source is the UK's national synchrotron science facility, located at the Harwell Science and Innovation Campus in Oxfordshire.
Copyright © 2022 Diamond Light Source
Diamond Light Source Ltd
Diamond House
Harwell Science & Innovation Campus
Didcot
Oxfordshire
OX11 0DE
Diamond Light Source® and the Diamond logo are registered trademarks of Diamond Light Source Ltd
Registered in England and Wales at Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United Kingdom. Company number: 4375679. VAT number: 287 461 957. Economic Operators Registration and Identification (EORI) number: GB287461957003.

 Science
Science